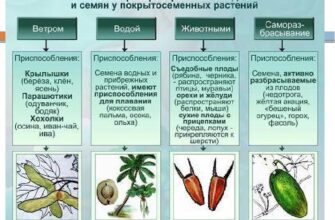
- Генетические нарушения у человека и методы их выявления
- Методы исследования хромосом
- Мутации в генах и заболевания, к которым они способны приводить
- Как выявляют рецессивные мутации?
- Что делать, если в семье есть наследственное заболевание?
- Научная электронная библиотека
- 4.2. Принципы клинической диагностики наследственных болезней
Генетические нарушения у человека и методы их выявления

Генами называются участки ДНК, в которых закодирована структура всех белков в теле человека или любого другого живого организма. В биологии действует правило: «один ген – один белок», то есть в каждом гене содержится информация только об одном определенном белке.
В 1990 году большая группа ученых из разных стран начала проект под названием «Геном человека». Он завершился в 2003 году и помог установить, что человеческий геном содержит 20–25 тысяч генов. Каждый ген представлен двумя копиями, которые кодируют один и тот же белок, но могут немного различаться. Большинство генов одинаковые у всех людей – различается всего 1%.
ДНК находится в клетке внутри ядра. Она особым образом организована в виде хромосом – эти нитеподобные структуры можно рассмотреть в микроскоп с достаточно большим увеличением. Внутри хромосомы ДНК намотана на белки – гистоны. Когда гены неактивны, они расположены очень компактно, а во время считывания генетического материала молекула ДНК расплетается.
В клетках человека есть структуры, которые называются митохондриями. Они выполняют роль «электростанций» и отвечают за дыхание. Это единственные клеточные органеллы, у которых есть собственная ДНК. И в ней тоже могут возникать нарушения. 
Весь набор хромосом в клетке называется кариотипом. В норме у человека он представлен 23 парами хромосом, всего их 46. Выделяют два вида хромосом:
- 22 пары аутосом одинаковы у мужчин и женщин. В каждой паре хромосомы имеют одинаковую длину и содержат одинаковые наборы генов.
- Одна пара половых хромосом. У женщин это две X-хромосомы. Одна из них неактивна и плотно свернута – ее называют тельцем Барра. У мужчин одна половая хромосома представлена X-хромосомой, а вторая – Y-хромосомой, она меньше по размерам.
Методы исследования хромосом
Для исследования кариотипа применяют специальный метод – световую микроскопию дифференциально окрашенных метафазных хромосом культивированных лимфоцитов периферической крови.
Этот анализ применяется для диагностики различных хромосомных заболеваний. Он позволяет выявлять такие нарушения, как:
- Грубые изменения в кариотипе – изменение количества хромосом. Например, при синдроме Дауна в клетках ребенка присутствует лишняя хромосома №21.
- Присутствие в организме клеток с разными кариотипами. Это явление называется мозаицизмом.
- Хромосомные аберрации – нарушение структуры хромосом, внутрихромосомные и межхромосомные перестройки. Сюда относятся делеции (утрата участка хромосомы), дупликации (удвоение участка хромосомы), инверсии (поворот участка хромосомы на 180 градусов), транслокации (перенос участка одной хромосомы в другую).

Однако с помощью исследования кариотипа можно выявить не все генетические нарушения. Оно не способно обнаружить такие изменения, как:
- микроделеции и микродупликации, когда утрачивается или дублируется очень маленький участок хромосомы;
- болезни обмена, вызванные нарушением последовательности «букв» генетического кода в отдельных генах;
- митохондриальные заболевания, связанные с нарушениями в генетическом материале митохондрий;
- низкопроцентный мозаицизм, когда клеток с неправильным кариотипом очень мало;
- мутации в отдельных генах, которые не приводят к изменению внешнего вида хромосом;
- эпигенетические расстройства, при которых структура хромосом и генов не меняется, но изменяется их функция.

Для получения дополнительной информации, не видимой в световой микроскоп, используют хромосомный микроматричный анализ (ХМА). С его помощью можно изучить все клинически значимые участки генома и выявить изменения в количестве и структуре хромосом, а именно микрополомки (микроделеции и микродупликации).
Во время хромосомного микроматричного анализа применяют технологию полногеномной амплификации и гибридизации фрагментов опытной ДНК с олигонуклеотидами, нанесенными на микроматрицу. Если объяснять простыми словами, то сначала ДНК, которую необходимо изучить, копируют, чтобы увеличить ее количество, а затем смешивают ее со специальными ДНК-микрочипами, которые помогают выявлять различные нарушения.
Эта методика позволяет в одном исследовании выявлять делеции и дупликации участков ДНК по всему геному. Разрешающая способность стандартного ХМА от 100 000 пар нуклеотидов – «букв» генетического кода (в отдельных регионах от 10 000 п. н.).
С помощью ХМА можно выявлять:
- изменения числа хромосом;
- дупликации и делеции, в том числе микродупликации и микроделеции;
- отсутствие гетерозиготности – утрату одной из двух копий гена. Это явление имеет важное значение в онкологии, при болезнях импринтинга (когда активность гена зависит от того, от какого из родителей он получен), аутосомно-рецессивных заболеваниях (связанных с рецессивными генами – о них мы поговорим ниже), близкородственных браках;
- однородительские дисомии, когда в геноме ребенка присутствуют две хромосомы от одного родителя.
Однако, как и предыдущий метод, хромосомный микроматричный анализ имеет некоторые ограничения. Он не позволяет выявлять или ограничен в выявлении таких аномалий, как:
- сбалансированные хромосомные аномалии, когда в хромосомах происходят изменения, которые не приводят к добавлению или утрате генетического материала. К ним относятся инверсии (разворот участка хромосомы на 180 градусов), реципрокные транслокации (обмен участками между хромосомами), небольшие инсерции (вставки в хромосомах);
- мозаицизм, если клеток с нарушениями в кариотипе менее 15%;
- CNV (copy number variation) – повторы небольших участков генома;
- точечные мутации – замены отдельных «букв» генетического кода;
- экспансия (увеличение) повторов коротких участков в ДНК;
- аномалии метилирования – присоединения особых метильных групп к определенным участкам ДНК, которые меняют активность генов.
Мутации в генах и заболевания, к которым они способны приводить
Мутации – это изменения, которые происходят в ДНК как случайным образом, так и под действием разных факторов, например химических веществ, ионизирующих излучений. Они могут затрагивать как отдельные «буквы» генетического кода, так и большие участки генома. Мутации происходят постоянно, и это основной двигатель эволюции. Чаще всего они бывают нейтральными, то есть ни на что не влияют, не приносят ни вреда, ни пользы. В редких случаях встречаются полезные мутации – они дают организму некоторые преимущества. Также встречаются вредные мутации – из-за них нарушается работа важных белков, наоборот, происходят достаточно часто. Генетические изменения, которые происходят более чем у 1% людей, называются полиморфизмами – это нормальная, естественная изменчивость ДНК Полиморфизмы ответственны за множество нормальных отличий между людьми, таких как цвет глаз, волос и группа крови.
Все внешние признаки и особенности работы организма, которые человек получает от родителей, передаются с помощью генов. Это важнейшее свойство всех живых организмов называется наследственностью. В зависимости от того, как проявляются гены в тех или иных признаках, их делят на две большие группы.
- Доминантные гены. Выражаясь простым языком, эти гены более «сильные». Если в клетках присутствует хотя бы одна копия такого гена, его признаки проявятся.
- Рецессивные гены «слабее» доминантных. Если у человека одна копия гена доминантная и одна рецессивная, – проявится признак доминантной. А для проявления рецессивного признака нужно две соответствующих копий.
Например, карий цвет глаз у человека является доминантным. Поэтому у кареглазых родителей с высокой вероятностью родится кареглазый ребенок. Если у одного из родителей глаза карие, а у другого голубые, то вероятность рождения кареглазых детей в такой семье тоже высока. У двух голубоглазых родителей, скорее всего, все дети тоже будут голубоглазыми. А вот у кареглазых родителей может родиться ребенок с голубыми глазами, если у обоих есть рецессивные «гены голубоглазости», и они достанутся ребенку. Конечно, это упрощенная схема, потому что за цвет глаз отвечает не один, а несколько генов, но на практике эти законы наследования зачастую работают. Аналогичным образом потомству могут передаваться и наследственные заболевания. 
Как выявляют рецессивные мутации?
Для выявления мутаций, которые передаются рецессивно, используют целый ряд исследований.
Секвенирование по Сэнгеру – метод секвенирования (определения последовательности нуклеотидов, буквально – «прочтение» генетического кода) ДНК, также известен как метод обрыва цепи. Анализ используется для подтверждения выявленных мутаций. Это лучший метод для идентификации коротких тандемных повторов и секвенирования отдельных генов. Метод может обрабатывать только относительно короткие последовательности ДНК (до 300–1000 пар оснований) одновременно. Однако самым большим недостатком этого метода является большое количество времени, которое требуется для его проведения.
Если неизвестно, какую нужно выявить мутацию, то используют специальные панели.
Панель исследования — тестирование на наличие определенных мутаций, входящих в перечень конкретной панели исследования. Анализ позволяет выявить одномоментно разные мутации, которые могут приводить к генетическим заболеваниям. Анализ позволяет компоновать мутации в панели по частоте встречаемости (скрининговые панели, направленные на выявление носительства патологической мутации, часто встречаемой в данном регионе или в определенной замкнутой популяции) и по поражаемому органу или системе органов (панель «Патология соединительной ткани»). Но и у этого анализа есть ограничения. Анализ не позволяет выявить хромосомные аберрации, мозаицизм и мутации, не включенные в панель, митохондриальные заболевания, а также эпигенетические нарушения.
Не в каждой семье можно отследить все возможные рецессивные заболевания. Тогда на помощь приходит секвенирование экзома – тест для определения генетических повреждений (мутаций) в ДНК путем исследования в одном тесте практически всех областей генома, кодирующих белки, изменения которых являются причиной наследственных болезней.
Секвенирование следующего поколения-NGS – определение последовательности нуклеотидов в геномной ДНК или в совокупности информационных РНК (транскриптоме) путем амплификации (копирования) множества коротких участков генов. Это разнообразие генных фрагментов в итоге покрывает всю совокупность целевых генов или, при необходимости, весь геном.
Анализ позволяет выявить точечные мутации, вставки, делеции, инверсии и перестановки в экзоме. Анализ не позволяет выявить большие перестройки; мутации с изменением числа копий (CNV); мутации, вовлеченные в трехаллельное наследование; мутации митохондриального генома; эпигенетические эффекты; большие тринуклеотидные повторы; рецессивные мутации, связанные с Х-хромосомой, у женщин при заболеваниях, связанных с неравномерной Х-деактивацией, фенокопии и однородительские дисомии, и гены, имеющие близкие по структуре псевдогены, могут не распознаваться. 
Что делать, если в семье есть наследственное заболевание?
Существуют два способа выявить наследственные генетические мутации у эмбриона:
Предимплантационное генетическое тестирование (ПГТ) в цикле ЭКО. Это диагностика генетических заболеваний у эмбриона человека перед имплантацией в слизистую оболочку матки, то есть до начала беременности. Обычно для анализа проводится биопсия одного бластомера (клетки зародыша) у эмбриона на стадии дробления (4–10 бластомеров). Существует несколько видов ПГТ: на хромосомные отклонения, на моногенные заболевания и на структурные хромосомные перестройки. Данные Simon с соавторами (2018) говорят о том, что в случае проведения ЭКО с ПГТ у пациентки 38–40 лет результативность ЭКО составляет 60%. Но при исследовании эмбриона есть ряд ограничений. Так, из-за ограниченного числа клеток можно не определить мозаицизм.
Если нет возможности провести ЭКО с ПГТ, то используют второй вариант – исследование плодного материала во время беременности.
Для забора плодного материала используют инвазивные методы:
- биопсия хориона – когда берут клетки из плаценты;
- амниоцентез – когда берут клетки амниотической жидкости.
Далее эти клетки исследуют при помощи одного или нескольких генетических тестов (которые имеют свои ограничения). Проведение инвазивных методов может быть связано с риском для беременности порядка 1%.
Таким образом, проведя дополнительные исследования, можно значительно снизить риск рождения ребенка с генетическим заболеванием в конкретной семье. Но привести этот риск к нулю на сегодняшний день, к сожалению, невозможно, так как любой генетический тест имеет ряд ограничений, что делает невозможным исключить абсолютно все генетические болезни.

Автор статьи
Пелина Ангелина Георгиевна
Ведёт генетическое обследование доноров Репробанка, осуществляет подбор доноров для пар, имеющих ранее рождённых детей с установленной генетической патологией.
Источник
Научная электронная библиотека

Юров И. Ю., Ворсанова С. Г., Воинова В. Ю., Чурносов М. И., Юров Ю. Б.,
4.2. Принципы клинической диагностики наследственных болезней
Диагностика наследственных заболеваний чрезвычайно сложна из-за их многочисленности (несколько тысяч нозологических форм) и низкой частоты (частота многих заболеваний составляет 1:200000 и более).
Термин синдром обычно применяют для обозначения совокупности воспроизводимых симптомов, имеющих общую этиологию. Однако в клинической генетике его используют для обозначения отдельных нозологических форм. Это связано с тем, что исторически многие из них были описаны как симптомокомплексы без понимания этиологии. Несмотря на то, что в дальнейшем стали известны генетические причины этих заболеваний, за ними традиционно сохранилось название «синдром». Таким образом, для наследственной патологии понятия «болезнь» и «синдром» равнозначны. Для обозначения некоторых нозологических форм употребляются оба термина (например, болезнь Дауна и синдром Дауна, болезнь Марфана и синдром Марфана).
Среди симптомов наследственных болезней редко встречаются патогномоничные. Чаще всего один и тот же признак встречается при различных заболеваниях. Так, катаракта и помутнение хрусталика наблюдаются при более чем 30 наследственных заболеваниях, а умственная отсталость при сотнях нозологических форм. Поэтому, в клинической генетике значим синдромальный подход к диагностике, согласно которому только сочетание симптомов является специфичным для конкретного заболевания.
На основе данных об особенностях клинических проявлений наследственной патологии строится схема обследования больного. Важным методом с точки зрения генетики является антропометрия. Необходимо оценивать следующие антропометрические показатели: длину и массу тела больного, тип телосложения, соотношение длины конечностей и туловища, а также отдельных частей конечностей (например, плеча и предплечья), окружности груди и головы, соотношение продольного и поперечного размера черепа, длину стопы и кисти. Эти данные оцениваются по специально разработанным центильным таблицам, основные из которых представлены в Приложении. При многих наследственных заболеваниях антропометрические показатели выходят за пределы допустимых вариаций вследствие нарушения роста скелета, диспропорциональности развития его отдельных частей. Например, высокий рост, длинные верхние и нижние конечности, пальцы могут быть признаками синдрома Марфана. Уменьшение окружности головы и размеров стоп помогают в диагностике синдрома Ретта.
Очень важно выявить у больного врождённые аномалии развития, которые могут быть либо проявлением наследственной патологии, либо следствием тератогенного воздействия средовых факторов. Следует остановиться на различиях между следующими употребляемыми в литературе терминами: врождённые пороки развития, врождённые аномалии, микроаномалии развития.
Врождённый порок развития – это морфологический дефект органа или его части, или большой области тела, ведущий к нарушению функции органа (органов) либо социальной адаптации индивидуума. Врождённая аномалия (или дефект) – более общее понятие, это любая функциональная или структурная аномалия, вызванная либо генетическим заболеванием, либо средовым событием, предшествующим рождению. Врождённые пороки развития подробно рассмотрены ниже (см раздел «Врождённые аномалии и синдромы», глава 6).
Микроаномалии развития (МАР) – необычные морфологические черты, которые, в отличие от пороков развития, не сопровождаются нарушениями функции органа или системы.
Как было сказано выше, при обследовании больных необходимо обращать внимание на МАР. Последние встречаются также и у здоровых людей, но наличие нескольких МАР указывает на высокую вероятность наследственной патологии. В таблицах 3 и 4 представлен перечень наиболее значимых микроаномалий и способы определения некоторых из них.
МАР органов или части тела
I. Лицевая область и остальная часть черепа
Лицо: плоское, круглое, треугольное, вытянутое, грубые черты, плоский профиль.
Брови: сросшиеся (синофриз), кустистые.
Нос: короткий, клювовидный, загнутый, седловидная переносица, широкая плоская переносица, плоские крылья носа, открытые вперёд ноздри.
Фильтр: длинный, короткий, плоский, глубокий.
Волосы: торчащие, два завитка (две макушки).
Голова: брахицефалия, долихоцефалия, тригоноцефалия, акроцефалия, выступающий или скошенный лоб, выступающий затылок, затылочная шпора (выступающая затылочная кость), плоский затылок.
II. Область глазных щелей и вокруг них
Веки: эпикант, телекант, птоз (малый), колобома век.
Глазные щели: узкие, короткие.
Разрез глазных щелей: монголоидный, антимонголоидный.
Расстояние между глазами: гипотелоризм, гипертелоризм.
Склеры: голубые, телеангиэктазии на склерах.
Радужная оболочка глаза: колобома, гетерохромия.
Ресницы: неправильный рост (например, двойной ряд ресниц).
III. Область ротовой полости и вокруг нее
Челюсти: прогения (выступающая нижняя челюсть), ретрогения (смещенная назад нижняя челюсть), прогнатия (выступающая верхняя челюсть), макрогения (большая нижняя челюсть) и микрогения (маленькая нижняя челюсть), микрогнатия (маленькая верхняя челюсть) и макрогнатия (большая верхняя челюсть).
Подбородок: скошенный, выступающий.
Язык: исчерченность, макроглоссия и микроглоссия (увеличение и уменьшение языка), короткая уздечка.
Губы: тонкие, толстые, с бороздами, с множественными уздечками.
Рот: макростомия (большой рот) и микростомия (маленький рот).
Нёбо: плоское, высокое, арковидное, готическое, раздвоение язычка.
Зубы: неправильное расположение, неправильная форма, врождённый избыток или врождённое отсутствие одного или нескольких зубов, гипоплазия эмали, диастема верхняя и нижняя (увеличенние промежутка между первыми резцами), тремы (широкие промежутки между зубами).
IV. Ушные раковины
Асимметрия размера, большие, маленькие оттопыренные, косо направленные, низко расположенные, приросшая мочка, маленькая или отсутствующая мочка, уплощенные ушные раковины, неполное развитие завитка со сглаженным упрощенным рисунком, отсутствие козелка, околоушные придатки, преаурикулярная фистула.
V. Верхние конечности
Длина пальцев: брахидактилия (короткие пальцы), укорочение отдельных пальцев, арахнодактилия (длинные «паучьи» пальцы).
Количество пальцев: полидактилия (дополнительные пальцы), олигодактилия (уменьшение количества пальцев).
Форма пальцев: утолщение ногтевых фаланг, конусовидная форма.
Подвижность пальцев: камптодактилия (тугоподвижность межфаланговых суставов), сверхгибкость (гиперэкстензия).
Мизинец: короткий, серповидный (клинодактилия), дополнительные складки, отсутствие сгибательной складки либо одна складка.
Большой палец: широкий, трёхфаланговый, его гипоплазия.
Ногти: удвоение ногтя большого пальца, гипоплазия ногтей.
Синдактилия (сросшиеся пальцы). Поперечная ладонная борозда (четырехпальцевая борозда).
VI. Нижние конечности
Форма и длина: укороченные, удлинённые, вальгусная деформация (Х-образные) или варусная деформация (О-образные).
Форма стопы: полая, стопа-качалка.
Пальцы стоп: синдактилия, асимметрия длины пальцев, 3-й палец длиннее 2-го, глубокая борозда между 1-м и 2-м пальцами, сандалевидная щель, широкий большой палец, полидактилия, брахидактилия.
Глубокая складка на стопе.
VII. Область шеи и туловища
Шея: короткая, длинная, кривошея, крыловидные складки, низкая линия роста волос на шее.
Грудная клетка: воронкообразная, килевидная, щитовидная.
Соски: гипертелоризм, резкая гипоплазия, расположение на разном уровне, полителия (добавочные соски).
Пилонидальная ямка (втяжение кожи в виде ямки, часто локализуется в области крестца).
VIII. Половые органы
Шалевидная мошонка, увеличенный клитор.
IX. Кожа, ее придатки и подкожная клетчатка
Пятна на коже: гемангиомы, телеангиэктазии, крупные невусы (пигментированные выступающие участки кожи диаметром более 1 см), волосатый невус, пигментация, депигментация.
Повышенная растяжимость кожи, «лишняя» кожа.
Волосы: сухие, редкие, шерстистые, седая прядь надо лбом, «мыс вдовы», низкий рост волос на лбу, гипертрихоз (избыточный рост волос), гирсутизм (повышенное оволосение у девочек по мужскому типу), алопеция (облысение тотальное, гнёздное).
Ногти: короткие, широкие, вогнутые, гипоплазия, дистрофия ногтей, ногти в виде «часовых стекол».
Необычные ямки на лице и туловище, липомы, фибромы.
МАР, часто исчезающие в течение первых 2 лет жизни следующие: плоская капиллярная гемангиома на лице и шее, неполное развитие завитка уха, гипоплазия ногтя большого пальца ноги, широкая переносица, эпикант.
Способы определения некоторых микроаномалий
Определяется у больного в положении лежа на спине – затылок, шея и спина располагаются на одном горизонтальном уровне.
Гипер- и гипотелоризм глазных щелей
Определяется с помощью орбитального индекса (Ио): отношение расстояния между внутренними углами глазных щелей к окружности головы на уровне глаз.
где а – расстояние между внутренними углами глазных щелей;
b – окружность головы на уровне глаз.
Ио ≥ 7,5 % – гипертелоризм, Ио ≤ 5,2 % – гипотелоризм.
Смещение внутренних углов глазных щелей латерально при нормально расположенных глазницах.
Косое расположение ушных раковин
Угол, образованный линиями, проходящими вдоль ушной раковины и вертикалью, проходящей через мочку, > 20°.
Асимметрия ушных раковин
Различия в размерах правого и левого уха ≥ 15 %.
Низкое расположение ушных раковин
Определяется при сопоставлении верхней точки прикрепления ушной раковины с уровнем латерального угла глаза.
Верхний его край совпадает или ниже уровня середины 2-й фаланги 4-го пальца.
Расстояние между 1-м и 2-м пальцами нижних конечностей ≥ ширины 2-го пальца.
Сращение пальцев > 1/3 длины одного пальца.
Вычисляется на основании соскового индекса (Си) – отношения расстояния между центрами сосков и окружностью груди на уровне сосков (в %).
Источник