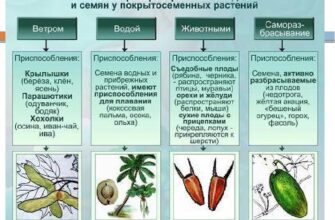
Публикации в СМИ
Дефекты репарации ДНК
Репарация — клеточный механизм коррекции повреждённой последовательности ДНК (точечные мутации, делеции, структурные нарушения и др.).
Системы репарации
• Темновая (фотореактивация): удаляются УФ-индуцированные ковалентные связи между смежными основаниями ДНК.
• Эксцизионная: удаляет неправильно спаренные или повреждённые основания из ДНК с последующим синтезом новой последовательности по неповреждённой цепи. Состоит из 5 этапов, контролируемых множеством генов, кодирующих участвующие в репарации ферменты (эндонуклеазы, экзонуклеазы, ДНК-полимераза, ДНК-лигаза): распознавание дефекта, рассечение повреждённого участка, удаление «ошибочного» олигонуклеотида, синтез ДНК с использованием комплементарной цепи как матрицы, лигирование. Дефекты эксцизионной репарации регистрируют при пигментной ксеродерме, синдроме Коккейна, трихотиодистрофии.
• Рекомбинационная: для восстановления одной хромосомы используется материал гомолога. Дефекты рекомбинационной репарации регистрируют при синдроме Блума.
Гены, участвующие в репарации ДНК • 126380, ген ERCC1, 19q13.2–19q13.3. До настоящего времени его клиническое значение неясно; совместно с белками XPA и XPF (продукт гена ERCC4) участвует в процессах распознавания и вырезания дефектного участка ДНК • 126340, ген ERCC2, 19q13.2–19q13. 3. Кодирует геликазу с неясными функциями; мутации вызывают пигментную ксеродерму типа D • 133510, ген ERCC3, 2q21. Кодирует геликазу репарации ДНК; различные мутации вызывают пигментную ксеродерму типа B, комплекс пигментной ксеродермы и синдрома Коккейна, трихотиодистрофию • 133530, ген ERCC5, 13q32–13q32, 13q33–13q33. Мутации вызывают комплекс пигментной ксеродермы и синдрома Коккейна типа А • 133540, ген ERCC6 (CKN2), 10q11–10q21 . Мутации вызывают синдром Коккейна типа В.
Приложение. Трихотиодистрофия — наследственное заболевание с ломкостью волос и ногтей (из-за уменьшенного содержания богатых цистеином матричных белков), ихтиозиформными изменениями кожи, физической и умственной отсталостью. Генетические аспекты: 50% пациентов имеют повышенную фоточувствительность, связанную с дефектом эксцизионной репарации ДНК (#601675, гены ERCC2/XPD и ERCC3/XPB, r ). Клиническая картина: ломкие волосы и ногти, ихтиоз, небуллёзная ихтиозиформная эритродермия, фоточувствительность, недоразвитие подкожной клетчатки, задержка физического развития, низкая масса плода при рождении, умственная отсталость, кишечная непроходимость, гипогонадизм, амастия, катаракта, микроцефалия, бронхиальная астма, контрактуры суставов, частые инфекционные заболевания. Лабораторные данные: снижение содержания богатого цистеином белка в волосах и ногтях, гипогаммаглобулинемия. Синонимы: синдром Тэя, врождённый ихтиоз с трихотиодистрофией. МКБ-10. L67.8 Другие аномалии цвета волос и волосяного стержня.
Код вставки на сайт
Дефекты репарации ДНК
Репарация — клеточный механизм коррекции повреждённой последовательности ДНК (точечные мутации, делеции, структурные нарушения и др.).
Системы репарации
• Темновая (фотореактивация): удаляются УФ-индуцированные ковалентные связи между смежными основаниями ДНК.
• Эксцизионная: удаляет неправильно спаренные или повреждённые основания из ДНК с последующим синтезом новой последовательности по неповреждённой цепи. Состоит из 5 этапов, контролируемых множеством генов, кодирующих участвующие в репарации ферменты (эндонуклеазы, экзонуклеазы, ДНК-полимераза, ДНК-лигаза): распознавание дефекта, рассечение повреждённого участка, удаление «ошибочного» олигонуклеотида, синтез ДНК с использованием комплементарной цепи как матрицы, лигирование. Дефекты эксцизионной репарации регистрируют при пигментной ксеродерме, синдроме Коккейна, трихотиодистрофии.
• Рекомбинационная: для восстановления одной хромосомы используется материал гомолога. Дефекты рекомбинационной репарации регистрируют при синдроме Блума.
Гены, участвующие в репарации ДНК • 126380, ген ERCC1, 19q13.2–19q13.3. До настоящего времени его клиническое значение неясно; совместно с белками XPA и XPF (продукт гена ERCC4) участвует в процессах распознавания и вырезания дефектного участка ДНК • 126340, ген ERCC2, 19q13.2–19q13. 3. Кодирует геликазу с неясными функциями; мутации вызывают пигментную ксеродерму типа D • 133510, ген ERCC3, 2q21. Кодирует геликазу репарации ДНК; различные мутации вызывают пигментную ксеродерму типа B, комплекс пигментной ксеродермы и синдрома Коккейна, трихотиодистрофию • 133530, ген ERCC5, 13q32–13q32, 13q33–13q33. Мутации вызывают комплекс пигментной ксеродермы и синдрома Коккейна типа А • 133540, ген ERCC6 (CKN2), 10q11–10q21 . Мутации вызывают синдром Коккейна типа В.
Приложение. Трихотиодистрофия — наследственное заболевание с ломкостью волос и ногтей (из-за уменьшенного содержания богатых цистеином матричных белков), ихтиозиформными изменениями кожи, физической и умственной отсталостью. Генетические аспекты: 50% пациентов имеют повышенную фоточувствительность, связанную с дефектом эксцизионной репарации ДНК (#601675, гены ERCC2/XPD и ERCC3/XPB, r ). Клиническая картина: ломкие волосы и ногти, ихтиоз, небуллёзная ихтиозиформная эритродермия, фоточувствительность, недоразвитие подкожной клетчатки, задержка физического развития, низкая масса плода при рождении, умственная отсталость, кишечная непроходимость, гипогонадизм, амастия, катаракта, микроцефалия, бронхиальная астма, контрактуры суставов, частые инфекционные заболевания. Лабораторные данные: снижение содержания богатого цистеином белка в волосах и ногтях, гипогаммаглобулинемия. Синонимы: синдром Тэя, врождённый ихтиоз с трихотиодистрофией. МКБ-10. L67.8 Другие аномалии цвета волос и волосяного стержня.
Источник
Способы репарации повреждений днк
Основные положения:
• Под влиянием факторов внешней среды или в результате ошибок в работе различных систем клетки в генетическом материале постоянно возникают повреждения
• Для сохранения жизнеспособности во всех клетках должны быть системы репарации, снижающие количество повреждений в ДНК
Наряду с безошибочным воспроизведением генетической информации важную роль играет поддержание ее информационной целостности. Фактически в геноме человека присутствует больше генов, ответственных за репарацию повреждений ДНК, чем кодирующих ферменты репликации.
Ошибки в последовательностях ДНК могут возникать по двум причинам. Во-первых, при репликации, во вновь образующуюся цепь ДНК может включиться неправильное основание. Для предотвращения таких ошибок в системе репликации существует корректорский механизм, который снижает число ошибочно включенных нуклеотидных остатков до минимума.
Во-вторых, при воздействии таких факторов внешней среды, как ионизирующие излучения и химические агенты, нарушающие структуру нуклеотидов, в ДНК могут возникнуть повреждения. В клетке существует много репаративных систем, которые устраняют повреждения в последовательностях ДНК, восстанавливая их правильную структуру.
Рисунок иллюстрирует действие репаративной системы, которая узнает повреждения в ДНК, удаляет их, и восстанавливает исходную структуру.
Несмотря на функционирование систем репарации, в генетическом материале возникают мутации, однако они не нарушают жизнедеятельность клетки. В действительности, определенная частота мутаций необходима для обеспечения вариабельности организмов в процессе эволюции.
Мутации во всех организмах, от бактерий до высших эукариот, возникают с частотой порядка 10 -6 в пересчете на один ген (или 10 -9 -10 -10 в пересчете на нуклеотидную пару) за одно поколение. Примерно такое же количество мутаций возникает даже у организмов, живущих в экстремальных условиях. Это позволяет предполагать, что общая частота возникновения мутаций определяется балансом между неблагоприятным эффектом большинства вредных мутаций и некоторыми полезными мутациями.
Ни одна клетка не может существовать в отсутствие систем репарации. Если, например, у Е. coli прекратить действие всех систем репарации, то однократное облучение бактерий УФ может оказаться летальным. В то же время бактерии с функционирующими системами репарации выносят огромное количество повреждений.
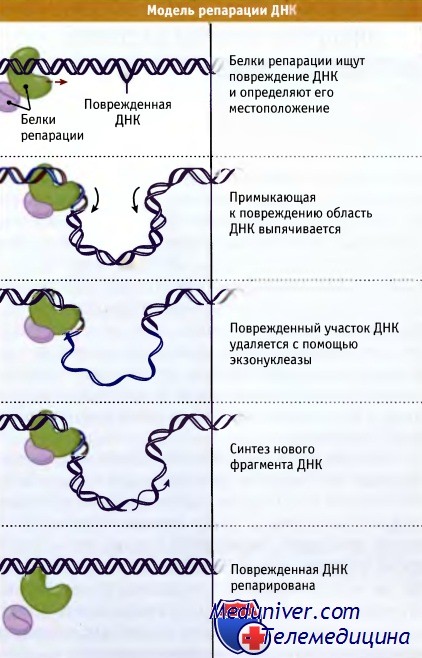 Система репарации узнает повреждение в ДНК,
Система репарации узнает повреждение в ДНК,
удаляет поврежденный участок и заполняет образующуюся брешь.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Источник
Материалы конгрессов и конференций
IX РОССИЙСКИЙ ОНКОЛОГИЧЕСКИЙ КОНГРЕСС
РЕПАРАЦИЯ ДНК И ЕЕ РОЛЬ В КАНЦЕРОГЕНЕЗЕ
Н.В. Томилин
Институт цитологии РАН, Санкт-Петербург
Много лет назад Джеймс Кливер и Дирк Бутсма показали, что фибробласты больных пигментной ксеродермой (ПК) имеют дефект по эксцизионной репарации (ЭР) ДНК после УФ облучения. Поскольку у большинства больных ПК образуется рак кожи, эти данные указывали, что у нормальных людей, редко болеющих раком кожи, репарация ДНК является важным фактором подавления канцерогенеза. По определению, репарация уменьшает число повреждений в ДНК, в том числе и вступающих в репликацию, тем самым, снижая вероятность образования мутаций и хромосомных перестроек, а, следовательно, инициацию рака и прогрессию опухолей. Эта простая концепция была впоследствии подтверждена на некоторых других моделях и стала весьма популярной, однако соотношения между репарацией ДНК и канцерогенезом оказались значительно более сложными, чем это представлялось ранее. Выяснилось, в частности, что до самой репарации работают сложные сигнальные каскады идентификации повреждений в геноме и выбора между репарацией и апоптозом, а также выбора адекватной системы репарации для данного типа повреждений. В последние годы значительный прогресс достигнут в понимании механизмов так называемой пострепликативной репарации или механизмов обхода повреждений ДНК во время и после репликации без их элиминации из генома, а также механизмов эпигеномных модификаций хроматина. Данный доклад будет посвящен краткому описанию известных к настоящему времени систем репарации ДНК в клетках высших эукариот и их роли в поддержании стабильности генома, обеспечивающей в нормальных клетках низкие темпы канцерогенеза.
Основной функцией ДНК, как известно, является сохранение наследственной информации путем полуконсервативной репликации. Матричный синтез, происходящий при репликации и транскрипции, следует правилам комплементарности азотистых оснований (А-Т и Г-Ц), основанным на их особой химической структуре, позволяющей ферментам этих синтезов, ДНК- и РНК-полимеразам, точно копировать последовательность нуклеотидов. Большинство этих ферментов строго различают нормальные звенья в матричных молекулах, поэтому, как правило, химические модификации нуклеотидов в ДНК приводят к блоку нормальной транскрипции и репликации, то есть являются некодирующими повреждениями. К таковым относятся внутритяжевые сшивки пиримидиновых нуклеотидов (циклобутановые димеры и 6-4 фотопродукты), образующиеся после УФ облучения, межтяжевые сшивки по пуриновым нуклеотидам, разнообразные ковалентные аддукты оснований, образуемые некоторыми химическими канцерогенами, тиминовые гликоли, апуриновые и апиримидиновые сайты и т.п. В редких случаях химические модификации нуклеотидов сохраняют способность к комплементарному спариванию во время репликации и транскрипции, однако это спаривание происходит неоднозначно. Например, окисленный гуанин, 8-оксогуанин (8-oxoG), спаривается не только с цитозином, но и с аденином, приводя к заменам в дочерних нитях ДНК при репликации. Можно отметить, что спонтанное окисление гуанина – это довольно частое событие, поскольку реакционно-способные соединения кислорода постоянно производятся митохондриями. И, наконец, к повреждениям можно отнести некоторые типы разрывов нитей ДНК, особенно при действии ионизирующих излучений, которые сопровождаются химическими модификациями оснований на концах.
Наиболее изученной системой репарации является эксцизионная репарация нуклеотидов (ЭРН), частично дефектная у большинства больных пигментной ксеродермой. При ЭРН модифицированные нуклеотиды (например, пиримидиновые димеры) удаляются из поврежденной нити благодаря действию нуклеаз XPG и ERCC1/XPF, а образующийся при этом однотяжевый пробел заполняется ДНК-полимеразами d или e с помощью PCNA и зашивается ДНК-лигазой. В ЭРН работают также геликазы XPD и XPB, входящие в состав комплекса TFIIH, и комплекс XPA/RPA, стабилизирующий расплетенный участок и, по-видимому, определяющий, в какой нити будут сделаны разрезы. Узнавание димеров в неактивной ДНК (так называемая глобальная репарация) обеспечивают комплексы XPC/HR23B и DDB2/DDB1, экспрессия которых контролируется хорошо известным опухолевым супрессором и транскрипционным фактором р53. Транскрипция гена DDB2 является индуцибельной, однако ген XPC экспрессируется постоянно.
При ЭРН повреждения ДНК могут распознаваться и без участия комплексов XPC/HR23B и DDB2/DDB1 при так называемой транскрипционной репарации, когда транскрипционный комплекс, содержащий РНК полимеразу II, блокируется на некодирующем повреждении, а репарация запускается белками CSA и CSB, дефектными при синдроме Кокейна. В отличие от больных с пигментной ксеродермой, у больных с синдромом Кокейна (рано стареющие кахектичные карлики) не наблюдается повышения частоты рака кожи, хотя фибробласты УФ-чувствительны. В одной из гипотез предполагается, что в клетках больных с синдромом Кокейна блок транскрипции быстро индуцирует апоптоз, поэтому раковые клоны образоваться не успевают.
Второй хорошо охарактеризованный путь репарации — это эксцизионная репарация оснований (ЭРО), при которой поврежденное азотистое основание, например тиминовый гликоль, удаляется специфической ДНК-гликозилазой до вскрытия сахарофосфатного каркаса ДНК. В настоящее время у млекопитающих идентифицированы 11 различных ДНК-гликозилаз (UNG, SMUG1, TDG, MBD4, MYH, OGG1, NTH1, NEIL1, NEIL2, NEIL3, MPG), некоторые из которых удаляют одни и те же повреждения. Например, 8-oxoG может удаляться из ДНК с помощью OGG1 и NEIL1, а дезаминированный цитозин (урацил) – с помощью UNG, SMUG1, TDG и MBD4. ДНК гликозилаза MYH удаляет только неспаренный аденин, вставляющийся при репликации в дочерние нити напротив 8-oxoG. Образующийся после действия ДНК-гликозилаз апуриновый или апиримидиновый сайт далее репарируется с помощью надрезания ДНК специфической нуклеазой APE1/REF1, ДНК-полимеразы β, вставляющей один неповрежденный нуклеотид, и комплекса XRCC4/LIG1 (short patch BER). В некоторых случаях комплексных повреждений во время ЭРО с помощью нуклеазы FEN1 удаляется несколько нуклеотидов, и тогда однотяжевый пробел заполняется с помощью PCNA и ДНК-полимеразы δ (long patch BER). У мышей инактивирующие гомозиготные мутации генов ДНК-полимеразы β, APE1/REF1, LIG1 и XRCC1 приводят к эмбриональной летальности, а мутации по ДНК-гликозилазам дают очень небольшие фенотипические отклонения, что может быть связано с параллелизмом их функций, а также с тем, что некоторые окисленные нуклеотиды инактивируются еще на уровне предшественников до их инсерции в ДНК. Например, 8-oxoGTP расщепляется до 8-oxoGMP фосфатазой MTH1, являющейся гомологом гена mutТ, одного из главных антимутаторов кишечной палочки.
Третий важный механизм репарации — это устранение неправильно спаренных нормальных нуклеотидов, гетеродуплексных пар или мизматчей, часто образующихся при репликации некоторых последовательностей в матричных нитях, например микросателлитов, а также при репликации обычных последовательностей нуклеотидов. Дефекты в генах системы репарации гетеродуплексов приводят к нестабильности микросателлитных сегментов и к мутациям по различным генам, в том числе и по генам опухолевых супрессоров. Почему-то особенно хорошо это проявляется в клетках эпителия толстого кишечника и приводит к высокой частоте наследственных неполипозных раков толстой кишки (HNPCC). Наиболее часто встречаются пациенты с мутациями генов hMLH1 (60% всех случаев HNPCC) и hMSH2 (35%), реже — hMSH6, а мутации по hPMS1 и hPMS2 обнаружены лишь в нескольких семьях. Мутации по генам hMLH3 и EXO1 (экзонуклеаза, взаимодействующая с hMSH2) тоже выявлены, но их значение пока неясно, поскольку у носителей рака нет. Мутаций по гену hMSH3 вообще пока не обнаружено.
Очень важной для клетки (и для канцерогенеза) является ее способность воссоединять случайные двойные разрывы (ДР) ДНК, что осуществляется двумя различными репарационными механизмами – негомогическим воссоединением концов (НГВК) ДНК и путем гомологической рекомбинации (ГР) при наличии по соседству второй копии неповрежденного идентичного по нуклеотидной последовательности сегмента ДНК, например сестринской хроматиды. Поскольку в диплоидных ядрах гомологичные хромосомы пространственно разделены (хромосомные территории), репарация путем ГР преимущественно происходит в S- и G2 фазах клеточного цикла, а НГВК осуществляется во время любой фазы цикла. В геномах высших эукариот имеется много повторов, по которым, в принципе, возможна репарация путем ГР, однако такая репарация ДР приводит к хромосомным транслокациям, и она практически полностью подавлена. Главным механизмом подавления потенциально кластогенной ГР между повторами и вообще неправильных (эктопических) воссоединений концов ДНК при репарации ДР является, по-видимому, локальная специфическая модификация хроматина по гистону Н2АХ – фосфорилирование лизина-139 (или образование γ-H2AX). Эта модификация, происходящая в мегабазных доменах хроматина рядом с ДР, способствует удержанию долгоживущих концов ДНК благодаря привлечению в эти домены специального белка когезинa (SMC1), а также белка hRad50, который может одномоментно связываться с двумя концами ДНК.
В клетках человека репарация путем НГВК непосредственно осуществляется продуктами генов DNA-PK, Ku70/80, XRCC4, LIG4, Artemis, hRAD50, hMRE11 и NBS1, а репарация путем ГР происходит с помощью белков RAD51 (paralogs XRCC2, XRCC3), RAD52, RAD54, BRCA1, BRCA2 (FANCD1), FANCD2, а также комплексов XPF/ERCC1 и hRAD50/hMRE11/NBS1. Узнавание случайных ДР осуществляется либо самим комплексом hRAD50/hMRE11/NBS1, который быстро связывается с концами ДНК, либо протеинкиназой АТМ (дефективной при наследственном синдроме атаксия-телеангиэктазия), которая выявляет какие-то не идентифицированные локальные изменения хроматина, вызванные ДР, и при этом активируется. АТМ-киназа быстро фосфорилирует гистон Н2АХ, белки NBS1, FANCD2 и Artemis, а также киназу Chk2 (RAD53), останавливающую клеточный цикл. Фосфорилирование белка Artemis необходимо для репарации примерно 10% случайных ДР. Образование γ-H2AX возможно и за счет активности других киназ (ATR и DNA-PK), но киназа ATR включается только при блоке репликации ДНК, когда в вилках репликации образуются достаточно протяженные однотяжевые бреши, связывающие белок RPA. Заметим, что репарация путем НГВК является неточной, и при ней всегда образуются микроделеции, тогда как ГР точно восстанавливает исходную последовательность нуклеотидов.
Важной для канцерогенеза является пострепликативная репарация (ПРР) или обход повреждений (lesion bypass or DNA damage tolerance) во время или после репликации. Этот путь был открыт у бактерий почти 40 лет назад, когда было показано, что напротив не вырезанных пиримидиновых димеров во время репликации образуются однотяжевые пробелы в дочерних нитях, которые затем устраняются гомологической рекомбинацией между сестринскими дуплексами ДНК, контролируемой белком RecA. ПРР рассматривается как главный источник индуцируемых мутаций.
Основные результаты по генному контролю ПРР в клетках эукариот были получены на дрожжах — это гены эпистатической группы RAD6 (RAD18, RAD5, MMS2, UBC13, RAD30, REV3, POL30). Гомологи большинства из этих генов идентифицированы в геномах высших эукариот. В настоящее время рассматриваются два основных (не альтернативных) механизма обхода некодирующих повреждений у эукариот: 1) прямой синтез ДНК напротив поврежденных нуклеотидов c помощью специальных ДНК-полимераз (translesion synthesis, TLS, or DNA polymerase switch); 2) использование как матрицы для синтеза напротив димера неповрежденного сестринского дуплекса без разрывов родительских нитей, смена матрицы (template switch, TMS). Небольшая часть пострепликативных пробелов может устраняться, по-видимому, и с помощью ГР между сестринскими дуплексами, при которой происходят разрывы родительских нитей ДНК и сестринские хроматидные обмены. Этот механизм ПРР аналогичен точной репарации двойных разрывов ДНК путем ГР, но касается только двойных разрывов, образующихся при репликации. При блоке репликации, однако, клетка стремится избежать рекомбинации, при которой образуются двойные разрывы ДНК в репликативных вилках. Подавление рекомбинации в заблокированных вилках происходит благодаря сумоилированию по лизину-164 белка PCNA ферментом UBC9, после чего SUMO-PCNA связывает геликазу SRS2, которая уже блокирует образование RAD51 филаментов. Дополнительными факторами подавления ГР в вилках репликации являются также известные геликазы BLM, WRN и RECQ4, дефективные при синдромах Блюма, Вернера и Ротмунда-Томсона, соответственно.
Главным событием при TLS является замена блокированной репликативной ДНК полимеразы (ДПδ или ДПε) на другую ДНК полимеразу, способную вставлять нормальные нуклеотиды напротив поврежденных звеньев. В клетках человека такой TLS-полимеразой является продукт гена, дефектного при вариантной пигментной ксеродерме, ДНК полимераза η (ДПη) (у дрожжей ген RAD30), которая связывается с репликативным белком PCNA (POL30), причем связывание усиливается при моно-убихитинилировании PCNA также по лизину-164. Эта модификация зависит от белка RAD18 и происходит при торможении репликации, однако остается неясным, каким образом при этом активируется RAD18. По нашим данным, при торможении репликации hRAD18, как и ДПη, накапливается в заблокированных вилках (фокусах репликации) и при этом перестает экстрагироваться из ядра буфером, содержащим Тритон-Х100. Эти изменения подавляются ингибиторами протеинкиназ стауроспорином и вортманнином, то есть могут быть связаны с фосфорилированием либо самого RAD18, либо какого-то взаимодействующего с ним белка. Есть основания предполагать, что это фосфорилирование осуществляется каскадом ATR/Chk1 в ответ на накопление однотяжевой ДНК в вилках репликации.
В дрожжах параллельно с моно-убихитинилированием PCNA происходит его поли-убихитинилирование по тому же лизину-164, зависисимое от продуктов генов RAD5, MMS2 и UBC13, которое направляет репарацию по второму механизму (смена матрицы, TMS). В клетках человека ПРР по механизму смены матрицы зависит от гомолога дрожжевого гена MMS2, однако поли-убихитинилирования PCNA пока не обнаружено. Необходимо отметить, что ПРР путем смены матрицы (TMS) является точной, а при смене полимеразы (TLS) специальные ДНК-полимеразы могут делать ошибки. Эти ошибки и являются, по-видимому, основным источником индуцированных точковых мутаций в геноме человека. Необходимо, однако, иметь ввиду, что только 5% генома кодирует белки и требует точного воспроизведения во время репликации, поэтому мутации в 95% генома (в некодирующей ДНК), особенно в дифференцированных соматических клетках, редко приводят к заметным фенотипическим эффектам. Что касается половых и стволовых клеток, то в них повреждения ДНК должны легче индуцировать апоптоз, а не репарацию.
Источник