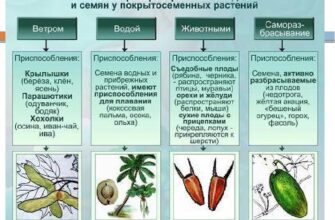
Методы разделения смесей и очистки веществ
Научные методы исследования химических веществ и превращений. Методы разделения смесей и очистки веществ.
Методы разделения смесей и очистки веществ
1. Разделение жидкостей
Перегонка – разделение жидкостей, различающихся по температуре кипения.
 | Перегонку проводят в приборе, состоящем из колбы Вюрца (или круглодонной колбы с газоотводной трубкой), прямого холодильника, колбы-приемника, аллонжа, термометра и нагревательного прибора. Смесь жидкостей нагревают в перегонной колбе до температуры кипения каждого компонента, пары отводят в холодильник и собирают сконденсировавшуюся жидкость в приемник, отдельно каждый компонент смеси. |
Дистилляция – очистка воды от твердых примесей.
 | Загрязненную жидкость нагревают в перегонной колбе до температуры кипения, пары отводят в холодильник и собирают сконденсировавшуюся жидкость в приемник. |
2. Разделение жидкостей и твердых веществ
Фильтрование – метод разделения жидкостей и твердых веществ.
 | Фильтрованием отделяют нерастворимые твердые вещества от жидкости. |
Выпаривание – метод разделения жидкостей и твердых веществ.
 | Выпариванием отделяют воду от растворенных в ней веществ. |
Декантация – с ливание жидкости с отстоявшегося осадка. К ней целесообразно прибегать в тех случаях, когда в сравнительно большом количестве жидкости находится немного твердого вещества, которое легко оседает на дно.
3. Разделение смесей твердых веществ
| Отстаивание | Магнитная сепарация |
| Например, смесь хлорида натрия и карбоната кальция можно разделить растворением в воде и последующим отстаиванием | Например, смесь серы и железных опилок можно разделить с помощью магнита: |


Сублимация (возгонка) — очистка твердых веществ, способных при нагревании переходить непосредственно из твердой фазы в газообразную, минуя жидкую фазу. Образующийся газ конденсируется охлаждаемой частью прибора. Сублимацию обычно проводят при температуре, близкой к температуре плавления вещества. Возгонкой можно очистить йод, серу, хлорид аммония.
Перекристаллизация. При повышенной температуре готовят насыщенный раствор очищаемого вещества, затем для удаления нерастворимых примесей раствор фильтруют через воронку для горячего фильтрования и охлаждают до низкой температуры. При понижении температуры растворимость вещества понижается, и основная часть очищаемого вещества выпадает в осадок, растворимые примеси остаются в растворе.
Хроматография. Метод разделения и анализа смесей веществ, который основан на распределении веществ между двумя фазами – неподвижной (твердая фаза или жидкость, связанная на носителе) и подвижной (газовая или жидкая фаза, элюент).
Источник
5.3. Анализ лекарственных смесей с разделением на компоненты
Разделение смесей на основании различной растворимости компонентов
Для разделения смесей лекарственных веществ на основе их различной растворимости в воде и органических растворителях используют разнообразные приемы.
Твердые лекарственные формы (порошки, таблетки) для разделения на ингредиенты обрабатывают соответствующим растворителем и фильтруют. В зависимости от дальнейшей методики анализа растворитель удаляют (отгоняют или выпаривают) или проводят определение в полученном растворе.
Примером могут служить смеси фенилсалицилата с гексаметилен-тетрамином и висмута нитратом основным.
ПРОПИСЬ 30 Фенилсалицилата
Гексаметилентетрамина по 0,25
Гексаметилентетрамин легко растворим в воде и спирте, растворим в хлороформе, очень мало растворим в эфире.
Фенилсалицилат практически нерастворим в воде, растворим в спирте и растворах щелочей, легко растворим в хлороформе, очень легко — в эфире.
Для количественного определения компонентов смеси навеску порошка взбалтывают с водой и фильтруют. Остаток промывают водой и определяют гексаметилентетрамин в фильтрате ацидиметрически.
Фенилсалицилат определяют в нерастворившемся остатке по общей методике анализа для сложных эфиров.
ПРОПИСЬ 31 Висмута нитрата основного Фенилсалицилата по 0,25
Висмута нитрат основной практически нерастворим в воде и спирте, легко растворим в кислотах азотной и хлороводородной. Растворимость фенилсалицилата — см. пропись 30.
Фенацетин очень мало растворим в воде, трудно растворим в ки-
пящей воде, растворим в спирте, мало растворим в эфире, хлороформе.
Навеску смеси обрабатывают водой, насыщенной фенацетином. Насыщенную фенацетином воду следует употреблять только после 24-часового настаивания, после чего в нее добавляют определенное количество натрия бензоата для лучшего растворения кофеина (в виде ко-феина-бензоата натрия). Смесь взбалтывают и фильтруют. Остаток на фильтре (фенацетин) растворяют в хлороформе, высушивают и определяют гравиметрическим методом.
Фильтрат подщелачивают раствором натрия гидроксида, экстрагируют из него кофеин хлороформом и определяют гравиметрически.
С помощью воды можно разделять и некоторые неорганические вещества (например, натрия гидрокарбонат и магния оксид). Многие из них также практически нерастворимы в органических растворителях (магния оксид, цинка сульфат, натрия тетраборат, висмута нитрат основной и др.) и при анализе с использованием эфира (хлороформа) остаются на фильтре. Примером служит пропись 36.
ПРОПИСЬ 36 Магния оксида
Натрия гидрокарбоната по 0,25
Магния оксид практически нерастворим в воде, свободной от углекислоты, и в спирте. Растворим в разведенных хлороводородной, сер-
ной, уксусной кислотах.
Натрия гидрокарбонат растворим в воде, нерастворим в спирте и эфире.
Навеску порошка взбалтывают с водой и фильтруют. Натрия гидрокарбонат количественно определяют в фильтрате ацидиметрически.
Магния оксид переносят с фильтра в колбу,.растворяют в 0,1 н. растворе кислоты хлороводородной, добавляют аммиачный буферный раствор и определяют лекарственное вещество комплексонометриче-ски.

Разделение смесей веществ, близких по растворимости, но отличающихся по кислотно-основным свойствам
Для разделения смесей лекарственных в^еств, растворимых в одних и тех же растворителях, но имеющих различные кислотно-основные свойства, используют химические реакции, в результате которых один из компонентов превращается в производное, обладающее иными
Чич еС \к,ркими свойствами и иной растворимостью. С этой целью чаще ^воДЯ^ут реакции нейтрализации или гидролиза.
Йзв^естно, что органические кислоты и основания при взаимодей-^ии с \ растворами щелочей или кислот переходят в соответствующие ‘^ и ‘ ВС \ следствие чего меняется их растворимость в органических рас-Аритед^лях g связи с этим для разделения лекарственных смесей, со-^ ЖаШ \л их ве[ Ц ества I и HI, I и V, III и V групп (см. стр. 10), используют тр ак \-цию 0рГаническим растворителем из кислого или щелочного с твор^а
^ ак \$с как для анализа лекарственных веществ I и III групп часто ис-
ilb 3 ^’ K \ симального разделения компонентов можно достигнуть, доба-точ.а подавления гидролиза солей избыток титранта после достиже-^крет*к и эквивалентности. Для уменьшения ошибок при определении 4 твор тного лекарственного вещества, остающегося в органическом раст Ъителе, необходимо тщательно разделять несмешивающиеся фа-xpaji-ворителей и органический слой тщательно промывать водой до Ан тьной реакции, выпк» :ализ смеси кислоты ацетилсалициловой и кофеина иллюстриру-\есказанное.
Ш ‘ОПИСЬ 37 Кислоты ацетилсалициловой 0,025 Кофеина 0,05
°Р°Ф\ л разделения ингредиентов используют извлечение кофеина ь ормом из щелочного раствора, в котором остается натриевая нтов \слоты ацетилсалициловой. Предварительное разделение компо-7 лени данной прописи необходимо, так как йодометрическому опре-л Д^К) кофеина мешает кислота ацетилсалициловая. ^/Щаюу $ определения кислоты ацетилсалициловой навеску смеси по-I , пли в делительную воронку, прибавляют воду, хлороформ, 2-3 щ* п — раствора фенолфталеина и быстро титруют, хорошо взбалтывая, I Ьаствором натрия гидроксида. Быстрое титрование необходимо, лт \ предотвратить гидролиз сложно-эфирной группы, а хорошее Чгвание — для полного перевода кислоты ацетилсалициловой в V фазу в виде натриевой соли. После достижения точки эквива-
лентности добавляют несколько избыточных капель титрованного раствора щелочи для предотвращения гидролиза натриевой соли кислоты ацетилсалициловой и хлороформный слой отделяют от водного (химизм реакции кислоты ацетилсалициловой и натрия гидроксида см. пропись 27).
Для определения кофеина хлороформ отгоняют и определяют лекарственное вещество в сухом остатке йодометрически:
Определение кофеина (как азотистого основания) в смеси с лекарственным веществом нейтрального характера (фенацетином) можно рассмотреть на примере прописи 35 (сравните обе методики). Компоненты смеси обладают примерно одинаковой растворимостью в воде и органических растворителях. При этом кофеин является слабым основанием, а фенацетин обладает нейтральным характером. Для разделения компонентов фенацетин подвергают кислотному гидролизу, в результате которого образуется и-фенететидин. Последний, обладая основными свойствами, образует соль, растворимую в воде и нерастворимую в органических растворителях.
Для определения кофеина навеску смеси кипятят с раствором кислоты хлороводородной и кофеин извлекают хлороформом. После отгонки растворителя кофеин определяют йодометрически (химизм — см. пропись 37) или ацидиметрически в неводной среде:
Подкисленный водный раствор, оставшийся после извлечения кофеина содержит гидрохлорид л-фенетидина, который может быть определен нитритометрически (химизм — см. пропись 16 на примере новокаина).
Разделение лекарственных веществ в мягких лекарственных формах
Для идентификации и количественного определения ингредиентов мягких лекарственных форм в экспресс-анализе используют методы, позволяющие проводить исследование в присутствии основы. В ряде случаев определяемое вещество приходится извлекать подходящим растворителем, чтобы освободиться от основы, мешающей анализу.
Основа в зависимости от природы может оказывать активное действие на скорость и полноту высвобождения лекарственного вещества.
Животные жиры (ланолин), растительные масла (масло какао, подсолнечное, персиковое и другие косточковые масла), углеводороды (вазелин, вазелиновое масло) относятся к числу гидрофобных основ. Они не растворяются в воде и этаноле, но хорошо растворяются в неполярных растворителях (хлороформ, эфир, гексан, бензол).
Для достижения полноты отделения лекарственного вещества важно правильно подобрать растворитель, учитывая при этом свойства не только анализируемого вещества, но и основы. Резорцин, калия йодид, кислота борная, димедрол и другие водорастворимые вещества можно извлекать водой, тогда как анестезин, оксиды цинка, магния и другие лучше извлекать кислотой хлороводородной разведенной в виде соответствующих гидрохлоридов. Так поступают при извлечении анестезина и цинка оксида из мягкой лекарственной формы (пропись 38).
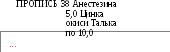 Методика. Анестезин. 0,5 г мази помещают в коническую колбу, добавляют 5 мл кислоты хлороводородной разведенной, 10 мл воды и нагревают на кипящей водяной бане в течение 5-7 минут, охлаждают и фильтруют через вату в коническую колбу. К фильтрату добавляют 0,5 г калия бромида, 2 капли раствора тропеолина 00, 1 каплю раствора метиленового синего и титруют 0,1 М раствором натрия нитрита при температуре 18 — 20° С, прибавляя его по 0,3 мл в начале титрования, а в конце титрования (за 0,2 мл до точки эквивалентности) — по одной капле до перехода красно-фиолетового окрашивания в голубое (химизм см. пропись 16 на примере новокаина).
Методика. Анестезин. 0,5 г мази помещают в коническую колбу, добавляют 5 мл кислоты хлороводородной разведенной, 10 мл воды и нагревают на кипящей водяной бане в течение 5-7 минут, охлаждают и фильтруют через вату в коническую колбу. К фильтрату добавляют 0,5 г калия бромида, 2 капли раствора тропеолина 00, 1 каплю раствора метиленового синего и титруют 0,1 М раствором натрия нитрита при температуре 18 — 20° С, прибавляя его по 0,3 мл в начале титрования, а в конце титрования (за 0,2 мл до точки эквивалентности) — по одной капле до перехода красно-фиолетового окрашивания в голубое (химизм см. пропись 16 на примере новокаина).
Параллельно проводят контрольный опыт на индикаторы. 1 мл 0,1 М раствора натрия нитрита соответствует 0,01652 г C9H11NO2 (анестезина).
NaN02 NaNO, /анестезин
Цинка окись, 0,5 г мази помещают в коническую колбу, прибавляют 5 мл кислоты хлороводородной разведенной, 10 мл воды, кипятят на водяной бане в течение 10 минут, охлаждают и фильтруют через вату в коническую колбу. Фильтрат нейтрализуют раствором аммиака (индикатор метиловый красный), прибавляют 5 — 7 мл аммиачного буферного раствора, 0,05 г индикаторной смеси кислотного хром-черного специального и титруют 0,05 М раствором трилона Б до появления синего окрашивания.
Тут вы можете оставить комментарий к выбранному абзацу или сообщить об ошибке.
Источник