- Большая Энциклопедия Нефти и Газа
- Получение — связанный азот
- Большая Энциклопедия Нефти и Газа
- Получение — связанный азот
- Технология связанного азота. Получение синтез-газа в промышленности.
- Сырьевая база азотной промышленности
- Получение синтез-газа из твердых топлив
- Получение синтез-газа из жидких углеводородов
- Получение синтез-газ из природного газа
- Сероочистка природного газа
- Каталитическая конверсия очищенного природного газа водяным паром
- Паровоздушная конверсия метана
- Конверсия оксида углерода водяным паром
- Катализаторы конверсии метана
Большая Энциклопедия Нефти и Газа
Получение — связанный азот
Получение связанного азота из атмосферного воздуха в плазменных реакторах интенсивно исследуется как у нас в стране, так и за рубежом, особенно в последние 10 лет. Пока плазменный метод по всем показателям уступает аммиачному, в первую очередь по расходу электроэнергии, который примерно в 7 — 10 раз выше. Однако разница становится менее ощутимой, если плазменный процесс совмещают с разложением фосфорсодержащего сырья в атмосфере воздуха с одновременной фиксацией азота. Дальнейшая переработка дает возможность получать из пятиокиси фосфора и окислов азота смесь фосфорной и азотной кислот для производства комплексных удобрений. Открываются определенные перспективы и для утилизации других компонентов фосфорсодержащего сырья. При диссоциации фосфорсодержащего сырья в плазме происходит практически полное его обесфторивание и выделение четырехфтористого кремния. Кроме того, отпадает необходимость в переработке фосфогипса, как это имеет место при сернокислотной переработке фосфатов, поскольку в плазмохимиче-ском процессе образуется окись кальция. [1]
Способ получения связанного азота в виде цианамида кальция был принят в различных странах с невероятной быстротой. [2]
Считается перспективным получение связанного азота с помощью солнечной энергии. [3]
Плазменные установки получения связанного азота имеют преимущества перед аммиачными. Эти установки компактны, независимы от источников сырья ( сырьем является воздух), процесс получения связанного азота является одностадийным. [4]
Существует три метода получения связанного азота : дуговой, цианамидный и аммиачный. [5]
Другой природный источник получения связанного азота — электрические разряды в атмосфере ( во время грозы), при которых образуются соединения азота. С дождевой водой они попадают в землю и образуют там соли азотной кислоты. [6]
Казалось бы, теперь проблема получения связанного азота решена. Но в действительности из-за огромного расхода электроэнергии на создание плазмы новый метод пока не имеет экономических преимуществ по сравнению с аммиачным. [7]
В настоящее время дуговой способ получения связанного азота быстро выходит из употребления. Даже и на больших норвежских заводах гидроэлектрическая энергия идет на производство электролитического водорода для синтеза аммиака. Проектное о-во Най-троджен ( Nitrogen Engineering Co. Норск-Гидро первую аммиачную установку этого типа в 1927 — 28 г., которая должна заменить старые дуговые установки. Эта установка в Но-тоддене была рассчитана на 5 300 тонн связанного азота в год, но благодаря добавочным количествам водорода первоначальная производительность была увеличена почти вдвое. Между тем в 1927 г. общество BASF и Норск-Гидро еще раз объединились для совместного производства связанного азрта и постройки большого аммиачного завода в Рьюкане. [8]
Подводя итоги, можно сказать, что проблема получения связанного азота в неограниченных количествах близится к разрешению. [9]
Так как исходными веществами этого широко распространенного метода получения связанного азота являются азот и водород, необходимо ознакомиться со способами их получения. [10]
Так как исходными веществами этого широко распространенного метода получения связанного азота являются азот и водород, необходимо ознакомиться со способами их получения. [11]
В начале нашего столетия применялись элсктродуговой циакамидный способы, получения связанного азота . [12]
В настоящее время синтез аммиака является основным промышленным методом получения связанного азота . [13]
В плазмотроне имеется возможность проведения такой эндотермической реакции, как, например, получение связанного азота из газов атмосферы по реакции N2 02 2NO — Q. Решение этой задачи позволит отказаться от сложного многостадийного и дорогого способа получения азотной кислоты из аммиака, значительно удешевит производство азотных удобрений. [14]
Наиболее перспективным методом переработки природного газа и других углеводородов в плазме является комплексная переработка с получением связанного азота , ацетилена и водорода. В результате проведенных исследований установлено, что при атмосферном давлении и температуре 2000 К пиролизом метана в азотной плазме можно получить одновременно до 10 5 % цианистого водорода и до 13 5 % ацетилена. Получение в больших количествах синильной кислоты без использования аммиака и дорогостоящих катализаторов ( платиновых, пла-тинородиевых) с одновременным получением ацетилена и водорода имеет важное значение, так как в этом случае стоимость продуктов связанного азота незначительна. Полученная плазменным методом синильная кислота может быть испольвована для производства высококачественных удобрений и дефолианта, пластмасс и других ценных продуктов. [15]
Источник
Большая Энциклопедия Нефти и Газа
Получение — связанный азот
Решение проблемы каучука так же, как в свое время разрешение важнейшей задачи о синтезе красителей и получении связанного азота из воздуха, является яркой демонстрацией могущества современной синтетической химии. [16]
За период с 1900 по 1920 г. делались и различные другие попытки разработать удовлетворительный в коммерческом отношении способ получения связанного азота , основанный на образовании цианида, но ни одна из них не имела сколько-нибудь значительного успеха. [17]
Примером технологического использования процесса горения топлива под давлением может служить также одна из первоочередных задач современной технической химии — получение связанного азота непосредственно из воздуха. [18]
Основным препятствием на пути увеличения объема производства азотных удобрений является их сравнительно высокая стоимость, которая обусловлена технологическими трудностями современных способов получения связанного азота . В настоящее время 85 % всего связанного азота в мире производится на основе гетерогенно-каталити-ческого синтеза аммиака из азота и водорода, который осуществляется при температуре 300 — 600 С и давлении 100 — 1000 атм. Такие условия, естественно, значительно удорожают технологию производства, и поэтому поиск более производительных и экономичных методов фиксации атмосферного азота является актуальнейшей задачей современной химической науки. [19]
Большой успех, выпавший на долю синтетического способа производства аммиака в Германии во время мировой войны, стимулировал всякого рода исследования, касающиеся этого способа получения связанного азота . В результате этих исследований, начатых во время войны во многих странах и продолжавшихся значительно более энергично непосредственно по ее окончании, был внесен целый ряд изменений в первоначальную схему Габер-Боша. По существу каждое из этих изменений является способом получения аммиака из его элементов, отличающимся от способа Габер-Боша в одном или в нескольких из нижеследующих отношений: а) источник водорода, б) способ отделения кислорода от азота воздуха, в) способ очистки водорода и азота, г) давления, температуры, д) способ отвода аммиака из системы, е) катализаторы. [20]
Работы Адольфа Франка, Каро, Роте, Фрейденберга, Менера и Альберта Франка по получению цианистого кальция уже упоминались в разделе, касающемся развития способа получения связанного азота в виде цианамида кальция. Как было указано выше, при обработке азотом карбида кальция образуется цианамид кальция в практически применимых условиях. При последующей плавке цианамида кальция с флюсом вроде обыкновенной соли получается продукт, содержащий цианистый кальций. Этот способ косвенного получения цианида применяется в настоящее время в промышленности. [21]
Сырьем являются газообразные или парообразные вещества, влектронагреваемае твердые частицы служат генераторами тепла и, в некоторых случаях, одним из реагентов Типичными представителями этой групш являются процессы пиролиза получения связанного азота , водяного газа. [22]
За четверть века, с 1913 г. ( когда был разработан метод синтеза аммиака) по 1938 г., ежегодное мировое производство связанного азота таким методом возросло от 7 до 1700 тыс. г. В настоящее время синтез аммиака является основным промышленным методом получения связанного азота . [24]
В результате этого процесса получали газы, содержащие 2 — 3 % окиси азота NO, которая в дальнейшем перерабатывалась в азотную кислоту. Получение связанного азота таким способом требовало затраты большого количества электроэнергии и могло быть осуществлено лишь на базе дешевой гидроэлектрической энергии. [25]
Энергетические затраты являются одним из главных показателей, определяющих экономику производства связанного азота плазмохимическим методом. Следует указать, что плазменный метод получения связанного азота является наиболее новым и недостаточно изученным. [26]
Использование селитры потеряло присущий ей когда-то характер гегемонии в получении связанного азота и идет на убыль. Среди новейших изысканий по азотным проблемам весьма многообещающим является синтез А. Эта задача, являющаяся задачей энергетич. [27]
Эти два основные природные источника получения азотных удобрений: селитра и каменный уголь — постепенно все же уменьшаются, а потребность в азотных удобр & ниях, наоборот, все более увеличивается. Поэтому еще в конце прошлого века встал вопрос об отыскании нового источника для получения связанного азота , причем, естественно, внимание ученых было обращено на колоссальные запасы свободного азота, содержащиеся в воздухе. После больших исследовательских работ вопрос этот был разрешен в начале текущего XX века. [28]
Плазменные установки получения связанного азота имеют преимущества перед аммиачными. Эти установки компактны, независимы от источников сырья ( сырьем является воздух), процесс получения связанного азота является одностадийным. [29]
Мировая война вызвала спрос на связанный азот, превзошедший все ожидания. Так как дуговой способ получения связанного азота нуждается в очень больших количествах энергии, то оказалось невозможным применять его где-либо, кроме как на нескольких установках незначительного масштаба. [30]
Источник
Технология связанного азота. Получение синтез-газа в промышленности.
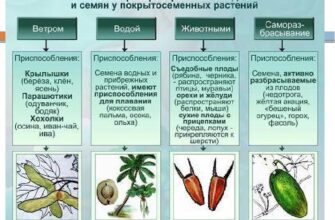
Азот и его соединения играют огромную роль в жизнедеятельности на Земле, так как он входит в состав аминокислот и белков.
Однако газообразный азот является очень устойчивым веществом и не обладает практически реакционной способностью (энергия связи N-N равна 945 кДж/моль, он обладает одной из самых высоких энтропий в расчете на атом).
Запасы азота в атмосфере огромные. Так, на 1 га поверхности Земли приходится около 80 тысяч тонн газообразного азота.
Некоторые количества азота связываются клубнями некоторых растений. Реакция катализируется ферментами, а необходимую энергию обеспечивает фотосинтез.
Некоторое количество газообразного азота из атмосферы связывается в результате грозовых разрядов по реакции:
N2 + O2 → 2 NO — Q;
Живым организмам требуется азот с валентностью -3, это аминокислоты и их полимеры — белки.
Но естественный процесс превращения азота в данную форму является очень долгим, и таким путем половина необходимого для жизнедеятельности на Земле количества азота может быть обеспечена только в течение 100 миллионов лет (для О2 в течение 3000 лет, для С в течение 100 лет).
Отсюда следует крайняя необходимость искусственного получения азотосодержащих соединений для использования их организмами.
Азотные соединения давно используются в фармакологии, военном деле, промышленности и с/х. Однако они были довольно дорогостоящими и вырабатывались в малых объемах. Природные месторождения азотных соединений (норвежская селитра — NaNО3 и чилийская селитра КNО3) в настоящее время практически выработаны.
Подлинной революцией в применении азотных соединений явилось открытие каталитической реакции синтеза аммиака из N2 и H2.
Сырьевая база азотной промышленности
Сырьем для получения продуктов в азотной промышленности служат азот из атмосферного воздуха, и газообразный водород, получаемый из различных видов топлива (твердого, жидкого, газообразного).
Так как запасы атмосферного воздуха, и, соответственно, содержащегося в нем азота, огромны, то сырьевая база азотной промышленности в основном определяется топливом, используемым для получения водорода или водородосодержащего газа. Для этих целей раннее широко использовалось твердое топливо (уголь) и коксовый газ.
В настоящее время для получения аммиака в основном используют природный газ (метан). В меньшем объеме используется попутный газ, коксовый газ, а также нефть.
Однако по мере истощения запасов природного газа и нефти вновь на первый план выйдет твердое топливо (путем предварительной газификации его водяным паром).
Смесь газов, которые служат сырьем для синтеза большого количества веществ, называются синтез-газами. Например, в производстве аммиака, метанола и др.
Получение синтез-газа из твердых топлив
Газификация твердых топлив — это один из наиболее старых видов производства. Его проводят в так называемых газогенераторах водяного газа. Сущность его заключается в попеременном пропускании через слой крупнокускового раскаленного твердого топлива воздушного и парового дутья.
Необходимая температура процесса 1000-1100 °C достигается путем частичного сжигания угля при продувании воздухом (реакция 1).
Синтез-газ образуется по эндотермической реакции (2). При этом температура снижается до 700-800 °C.
Циклы сменяются через 3-5 мин. Образующийся синтез-газ содержит 50-53 % Н2 и 36 % СО.
Для дальнейшей переработки синтез-газа прежде всего требуется очистить его сернистых примесей. А затем провести конверсию СО в СО2: Это достигается за счет каталитического взаимодействия СО с водяным паром по реакции:
Образующийся CO2 легко удаляется при помощи абсорбции каким-либо жидким поглотителем.
Недостатки данного процесса заключаются в его периодичности, низкой производительности газогенераторов, а также в высоких требованиях к качеству перерабатываемого угля (химсоставу, грансоставу).
В настоящее время разрабатываются более современные газогенераторы непрерывного действия кипящего слоя. В будущем это будет наиболее перспективным способом получения синтез-газа.
Получение синтез-газа из жидких углеводородов
В Японии и ФРГ более 60 % всего количества аммиака получают таким способом.
Для этих целей могут быть использованы как тяжелые жидкие топлива (мазут, крекинг-остатки и др.), так и легкие нефтепродукты, имеющие tкип не выше 200-220 °C (бензин, светлые дистилляты).
В первом случае тяжелые топлива под давлением (15 МПа) подогревается до t = 400-600 °C подается в газогенератор. Туда же подают подогретый кислород и перегретый водяной пар. При температуре 1350-1450 °C образуется синтез-газ, однако при этом образуется также некоторое количество сажи.
Газ очищают от сажи и сернистых соединений. Он имеет следующий состав: 3-5% CO2; 45-48 % СО, 40-45 % Н2, а также определенное количество метана, азота и аргона, проходит конверсию СО и очистку от СО2. Процесс протекает под давлением, которое может достигать 15 МПа. Производительность агрегатов до 30 тыс. м 3 /ч (H2+CO) и более.
Достоинства: возможность использования малоценного топлива.
Недостатки: высокий расход кислорода, выделение сажи, сложность технологической схемы.
Во втором случае легкие топлива перезабываются каталитически с водяным паром. На первой стадии топливо под давлением испаряется в подогревателе и тщательно очищается от примесей (катализаторных ядов). Так, содержание сернистых соединений не должно превышать 1 мг/кг. Далее пары смешиваются с перегретым водяным паром и смесь подается в реакционные трубы трубчатой печи, заполненные никелевым катализатором.
Этот процесс разработанный в 60-е годы, широко используется за рубежом.
Достоинства: меньший расход э/энергии, лёгкость регулирования, меньшие температуры.
Недостатки: высокие требования по очистке топлива от катализаторных ядов и большой удельный расход топлива.
Получение синтез-газ из природного газа
Основным сырьем для получения аммиака и метанола в нашей стране в настоящее время являются углеводородные газы (природный газ и реже попутный газ).
На рисунке представлена блок-схема получения синтез-газа из природного газа в производстве аммиака.

Сероочистка природного газа
Сернистые примеси, содержащиеся в природном газе зависимости от типа месторождения в количествах от 5 до 450 мг/м 3 , являются сильными катализаторными ядами. Кроме того, они вызывают интенсивную коррозию оборудования.
В настоящее время известно более 30 различных методов очистки от сернистых примесей, которые можно подразделить на три группы: хемосорбционно-каталитические; адсорбционные; и абсорбционные. При содержании серы в исходном газе до 80 мг/м 3 наиболее экономичным являются хемосорбционно-каталитические.
Природный газ наиболее легко очищается этим методом от примесей H2S. Дисульфиды и тяжелые меркаптаны отделяются гораздо труднее. По этой причине сероочистку проводят в две стадии:
1) На первой стадии сераорганические примеси предварительно гидрируют с использованием AL-Co-Mo или Al-Ni-Mo катализатора при температуре 350-400 °C и давлении 2-4 МПа.
Эти реакции в присутствии большого избытка Н2 практически необратимы, благодаря чему достигается практическое полное гидрирование трудно удалимых сернистых примесей.
Б) На второй стадии образовавшийся сероводород при температуре 390-410 °C легко поглощается твердым поглотителем на основе ZnO (например, марка ГИАП-10)
Эта реакция является также практически необратимой, что обеспечивает высокую степень улавливания Н2S. После достижения предельной сероемкости (20%) отработанный адсорбент выгружают и отправляют на переплавку.
После очистки содержание сернистых примесей в природном газе не должно превышать 0,5 мг/м 3 .
Кроме описанного метода, при повышенных концентрациях сернистых соединений применяют адсорбционные методы очистки на цеолитах (NaX-в состав которых входят Na2O; Al2O3; SiO2). Процесс адсорбции протекает при комнатной температуре, регенерация адсорбентов при 300-400 °C путем продувки азотом либо очищенным технологическим газом.
Каталитическая конверсия очищенного природного газа водяным паром
Паровая конверсия описывается реакцией:
При этом протекает и побочная нежелательная реакция крекинга:
Для подавления ее используется практически четырехкратный избыток водяного пара.
При Р = 0,1 МПа достаточно полная паровая конверсия метана достигается при температуре около 800 °C. Увеличение избытка водяного пара дополнительно увеличивает полноту конверсии.
Увеличение давления снижает полноту конверсии, так, при Р = 3 МПа требуемая температура составляет уже около 1100 °C.
В современных установках при Р = 2 МПа и выше, при соотношении СН4 : Н2Опар = 1 : 4, остаточное содержание метана после паровой конверсии составляет 8-10 %.
Для более полного превращения СН4 (≈ 0.5 %) конверсию ведут в две стадии:
— на первой стадии — паровая конверсия;
— на второй стадии — паровоздушная конверсия с использованием кислорода воздуха.
При этом кроме того в состав продуктов реакции вводится азот и получается синтез-газ практически стехиометрического состава, необходимого для последующего получения аммиака (N2 + 3H2 ⇔ 2NH3).
Паровоздушная конверсия метана
Это реакции неполного окисления метана. Они также протекают в две стадии.
1) При этом часть СН4 вступает в необратимые реакции:
СН4 + 0,5О2 = СО + 2Н2 + 347 кДж;
При этом температура достигает 1000 °C и более.
2) Оставшаяся часть метана вступает в каталитические реакции:
СН4 + СО2 ⇔ 2СО + 2Н2 — 248.1 кДж.
Первые реакции практически необратимы, поэтому повышение концентрации О2 сверх стехиометрического количества не дает дополнительного положительного эффекта. Повышение давления также термодинамически нецелесообразно.
Реакции паровой и углекислотной конверсии являются эндотермическими и требуют подвода тепла.
Реакции кислородной конверсии экзотермичны, причем выделяющейся теплоты достаточно для автотермического протекания паровой и углекислотной конверсии.
Конверсия оксида углерода водяным паром
Как видно из химических реакций при паровой конверсии образуется большее количество СО и его концентрация в конвертированном газе достигает 11 %. Для получения дополнительного количества водорода и снижения до минимума концентрации СО в конвертированном газ, осуществляется самостоятельная стадия — каталитическая конверсия СО водяным паром:
Реакцию можно сдвинуть вправо использованием избытка водяного пара, а также удалением СО2 из зоны реакции и снижением температуры. Давление на равновесие не влияет. Однако повышение давления экономически оправдано, т.к. при этом возрастает скорость реакции, уменьшаются требуемые габариты аппаратов и полезно используется энергия ранее сжатого природного газа.
Обычно концентрация паров воды в газе определяется количеством, дозируемым на конверсию СН4 и оставшимся после него Н2О: газ ≈ 0,4-0,5. Понижение температуры ограничено точкой росы (при Р = 2÷3 МПа, t росы =180-200 °C) и требует низкотемпературных катализаторов.
На практике чаще всего конверсию СО также проводят в две стадии:
— на первой стадии при относительно высокой температуре 400-500 °C обеспечивается высокая скорость конверсии большей части СО (катализатор среднетемпературный — железохромовый);
— на второй стадии при пониженной температуре 200-250 °C добиваются высокой степени превращения оставшегося количества СО (катализатор -низкотемпературный Ni, Zn, Al, иногда Gr). Выделяющееся тепло реакции используется для получения пара.
Температурный режим на каждой стадии зависит от свойств используемых катализаторов.
На первой стадии используют железохромовый катализатор в виде таблеток (среднетемпературный).
Ядами являются: S; Р; B; Si; Cl.
На второй стадии низкотемпературный — соединения меди, цинка, алюминия иногда хрома. В качестве добавок стабилизирующих характеристики катализатора, служат Al, Мg, Мn.
Перед эксплуатацией этот катализатор восстанавливают оксидом углерода или водородом. Срок службы меньше, чем среднетемпературных и не превышает 2-х лет. Концентрация Н2S должна быть не более 0,5 мг/ м 3 , ядом также является NH3.
Принципиальная технологическая схема агрегата конверсии природного газа мощностью 400000 т в год (1290 т/сутки) (по NH3)
Широкое применение в мировой и отечественной практике получили процессы каталитической двухступенчатой, паровой и паровоздушной конверсии, под давлением. При этом уменьшается расход на сжатие конвертированного газа, объем которого больше объема исходных газов, уменьшаются габариты аппаратов, полнее утилизируются тепло химических реакций, упрощается конструкция последующего компрессора для синтеза аммиака и создаются условия для сооружения агрегатов БЕМ с использованием энерготехнологических принципов.
На схеме приведена принципиальная технологическая схема двухступенчатой конверсии СН4 и СО под давлением агрегата большой единичной мощности (производительностью 1360 т/аммиака в сутки).

1 — компрессор природного газа; 2 — газовый подогреватель; 3 — реактор гидрирования сернистых примесей, 4 — адсорбер; 5 — дымосос; 6, 7, 9, 10 — подогреватели природного газа, питательной воды, паровоздушной парогазовой смесей, соответственно; 8 — пароперегреватель; 11 — реакционные трубы с катализатором; 12 — трубчатая печь паровой конверсии природного газа; 13 — шахтный конвертор природного газа второй степени; 14, 16 — паровые котлы; 15, 17 — конверторы СО первой и второй ступеней; 18 — теплообменник; 19 — компрессор воздуха
Природный газ сжимают в компрессоре 1 до давления 4,6 МПа, смешивают с азотоводородной смесью (авс) в соотношении авс : газ 1÷10 и подают в огневой подогреватель 2, где реакционная смесь нагревается от 130-140 °C до 370-400 °C. Для обогрева используется природный или другой газ.
Далее его подвергают двухступенчатой очистке от сернистых соединений: в реакторе 3 гидрируют на Al; Co; Mo -катализаторе, а затем образовавшийся Н2S адсорбируют в адсорбере 4 на сорбенте на основе ZnO. Остаточное содержание H2S в очищенном газе не должно превышать 0,5 мг/м 3 .
Очищенный газ смешивают с водяным паром в соотношении 1 : 3,7 и направляют в конвекционную камеру трубчатой печи 12. Далее парогазовая смесь поступает в радиантную камеру этой печи.
В радиационной камере размещены вертикальные трубы, заполненные никелевым катализатором конверсии метана и горелки, в которых сжигают топливный (природный) газ. Полученные в горелках дымовые газы обогревают трубы с катализатором.
Оставшееся тепло этих газов рекуперируют в конвекционной камере печи. Здесь размещены подогреватели парогазовой и паровоздушной смеси, перегреватели пара высокого давления, подогреватели питательной воды высокого давления и природного газа.
Парогазовая смесь нагревается в подогревателе 10 до 250 °C и затем под давлением 3,7 МПа распределяется сверху вниз по большому числу параллельных труб, заполненных Ni-катализатором. Выходящая из трубчатого реактора парогазовая смесь содержит 9-10 % CH4. При температуре 850 °C эта смесь поступает в шахтный конвертор второй ступени паровоздушной конверсии 13. Туда же в верхнюю часть компрессором 19 подается расчетное количество нагретой до 480-500 °C паровоздушной смеси. Парогазовая и паровоздушная смесь подаются в соотношении, которое обеспечивает практически полную конверсию остаточного количества CH4.
При температуре 1000-1100 °C продукты реакции направляются в котел-утилизатор 14, вырабатывающий пар давлением 10,5 МПа, где охлаждаются до 380-420 °C и поступает в конвертор СО первой ступени 15, где на среднетемпературном железохромовом катализаторе протекает конверсия основного количества СО водяным паром.
Начальное содержание СО в газе составляет 16-18 %, на выходе из реактора, оно снижается до 3,5-3,6 % СО, а температура 450 °C.
Далее газ подается в паровой котел 16, где также вырабатывается водяной пар с давлением 10,5 МПа, охлаждается до 225 °C и поступает в конвертор второй ступени 17, заполненный низкотемпературным катализатором. При этом остаточное содержание СО снижается до 0,5 % и газ имеет следующий состав: Н2 — 61,7 %; СО — 0,5%; CO2 — 17.4 %; N2+Ar — 20,1 %; CH4 — 0,3 %.
После дополнительного охлаждения в теплообменнике 18 до температуры окружающей среды и давлении 2,6 МПа газ поступает на очистку от СО2.
Получаемый пар Р = 10,5 МПа поступает на паровые турбины, приводящие в движение компрессоры и насосы производства аммиака.
Катализаторы конверсии метана
Для ускорения взаимодействия метана с водяным паром и диоксидом углерода используются катализаторы. При этом подавляется также побочная реакция:
Наибольшей активностью в данном процессе обладают никелевые катализаторы на инертном носителе — глиноземе (Al2О3). Они выпускаются в виде таблеток и колец Рашига. Например катализатор марки ГИАП-16 имеет следующий состав: 25% NiO; 57% Al2O3; 10 % СаО; 8% MgО.
Срок службы катализаторов конверсии метана более 3-х лет. Их активность снижается под воздействием ядов — сернистых соединений, а также углерода (закоксовывание). Отравление обратимо и после протекания чистого синтез-газа. При определенной температуре его активность практически полностью восстанавливается.
Закоксовывание можно устранить путем обработки катализатора водяным паром.
Источник