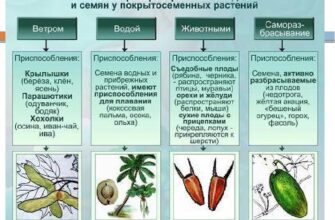
Способы получения дикарбоновых кислот
Дикарбоновыми кислотами называются производные углеводородов, содержащие две карбоксильные группы в составе молекулы. Химические и физические свойства дикарбоновых кислот сильно зависят от количества атомов в цепи, разделяющих карбоксильные группы. Так, кислотность возрастает при их сближении, т.к. карбоксилы оказывают друг на друга отрицательный индуктивный эффект. Те соединения, в которых карбоксильные группы разделены более чем двумя атомами углерода, напоминают по свойствам уксусную кислоту.

По официальной номенклатуре названия дикарбоновых кислот производят от названия соответствующего алкана, добавляя окончание «диовая кислота». Иначе, за основу берут алкан на два атома короче (без СООН-групп), и прибавляют окончание «дикарбоновая кислота», например:

Нумерация атомов цепи, как и для обычных кислот, проводится таким образом, чтобы карбоксильные группы получили наименьшие номера.
Основной способ получения щавелевой (этандиовой) кислоты заключается в пиролизе формиата натрия. При этом происходит гомолитический разрыв связи С-Н и последующая рекомбинация образующихся анион-радикалов.

Щавелевая кислота обладает более высокой кислотностью, чем уксусная. Это связано с акцепторным влиянием одной карбонильной группы на другую. Вторая константа ионизации уже не сильно отличается от обычных алифатических кислот, т.к. карбоксилатная группа акцепторными свойствами не обладает. Щавелевая кислота вступает в характерные для карбоновых кислот превращения, например, образует сложные эфиры и амиды.

Симметричный хлорангидрид щавелевой кислоты оксалилхлорид является весьма активным соединением, и имеет склонность к реакциям, сопровождающимся разрывом связи С-С. Его применяют для хлоркарбонилирования органических соединений.

Под действием сильных протонных кислот щавелевая кислота разлагается с образованием окиси и двуокиси углерода.

Малоновую (пропан-1,3-диовую) кислоту обычно синтезируют из хлоруксусной: сначала действием цианид-ионом ее переводят в циануксусную, затем проводят гидролиз, либо алкоголиз в кислой среде. В первом случае получается собственно малоновая кислота, а во втором – ее диэтиловый эфир.


Малоновая кислота и ее эфир обладают свойствами, которые присущи β-дикарбонильным соединениям, поэтому химия этих соединений более многогранна, нежели свойства простых кислот. Рассмотрим их поведение подробнее.
Свойства малоновой кислоты
Малоновая кислота и ее производные в большей степени, чем многие другие кислоты, склонна к декарбоксилированию и этим она сильно напоминает ацетоуксусную. Декарбоксилирование малоновой кислоты, как и ацетоуксусной, протекает по пути электроциклической реакции в шестичленном 6π-электронном переходном состоянии, в котором облегчена миграция электронных пар и элиминирование СО2.

Наличие β-дикарбонильного фрагмента в молекуле приводит к тому, что малоновая кислота может выступать не только как ОН-, но и как СН-кислота (образует енолят-анион). Это делает возможной электрофильную атаку α-углеродного атома в щелочной среде; например, для малоновой кислоты характерна конденсация с оксосоединениями, в первую очередь с ароматическими альдегидами – разновидность реакции Кневенагеля.

Образующаяся в результате арилиденмалоновая кислота неустойчива и очень легко декарбоксилируется, давая соответствующие коричные кислоты в качестве основных продуктов.
Свойства малонового эфира
Малоновый эфир, или диэтилмалонат способен под действием оснований давать исключительно енолят-анион, поэтому его активность по отношению к электрофилам выше. Сильные основания, такие как этилат натрия (магния), металлоорганические соединения и т.д., нацело депротонируют малоновый эфир. Полученные таким образом металлические производные по метиленовой группе находят широкое применение в синтезе, т.к. легко алкилируются и ацилируются а образующиеся при этом замещенные малоновые эфиры можно подвергать гидролизу и декарбоксилированию, получая разнообразные ценные продукты. Здесь просматривается прямая аналогия с синтезами на основе ацетоуксусного эфира (кетонное расщепление). См. главу Производные кислот, ацетоуксусный эфир.
Так, с помощью натриймалонового эфира можно синтезировать разнообразные карбоновые кислоты.


Если на первой стадии проводить ацилирование, то после расщепления могут быть получены кетоны сложного строения. Происходит двукратное декарбоксилирование, которое возможно благодаря тому, что вначале образуется β-кетокислота, которая, как известно, без труда декарбоксилируется при небольшом нагревании.

Для синтеза дикарбоновых кислот на первой стадии «сшивают» две молекулы натриймалонового эфира углеводородным мостиком, используя для этого алкилирование виц-дигалогеналканами.

Если требуется получить замещенные янтарные кислоты, на натриймалоновый эфир действуют молекулярным иодом. При этом сначала в результате сшивания метиновых С-атомов двух молекул натриймалонового эфира возникает эфир тетракарбоновой кислоты. Последний при гидролизе и декарбоксилировании дает янтарную кислоту.

Малоновый эфир, являясь 1,3-бисэлектрофилом, находит широкое применение и в синтезе гетероциклических соединений. Например, реакция с мочевиной в щелочной среде дает 2,4,6-пиримидинтрион – барбитуровую кислоту, которая ранее по своиму происхождению называлась малонилмочевиной.

Янтарную (бутандиовую) кислоту синтезируют по методологии, которая аналогична вышеизложенной для получения малоновой. В качестве исходного соединения используют 1,2-дибромэтан. Гидролиз нитрильных групп проводится в щелочной либо кислой среде.

Янтарная кислота проявляет характерные свойства карбоновых кислот – образует сложные эфиры со спиртами, амиды, хлорангидрид и другие функциональные производные.

Вместе с тем, четырехуглеродная цепь молекулы обусловливает определенные особенности химических свойств. Поскольку янтарная кислота является 1,4-бисэлектрофилом, ее взаимодействие с нуклеофилами может приводить к образованию циклов. Действительно, она весьма легко превращается в циклический ангидрид при обработке водоотнимающими средствами и даже при нагревании.

Янтарный ангидрид, как и другие ангидриды карбокислот, взаимодействует с нуклеофилами. Если ацилирование спиртов приводит к полным эфирам янтарной кислоты, то реакция янтарного ангидрида с аммиаком дает циклическое азотистое производное – сукцинимид, находящий широкое применение в тонком органическом синтезе.

Благодаря наличию двух электроноакцепторных карбонильных групп по соседству с атомом азота NH-связь в молекуле сукцинимида сильно поляризована, и легко диссоциирует под действием оснований. Образующийся при этом анион является активным нуклеофилом, и реагирует с разнообразными положительно заряженными частицами. Взаимодействие сукцинимида с галогенами (хлор, бром) приводит к соответствующим N-галогенпроизводным – N-бромсукцинимиду (NBS) и N-хлорсукцинимиду (NCS).

Эти соединения широко используются в органическом синтезе в качестве эффективных галогенирующих реагентов. NBS и NCS склонны к гомолитической диссоциации с высвобождением радикалов галогена, поэтому применяются именно для свободно-радикального галогенирования, в первую очередь, боковых цепей ароматических соединений, а также α-углеродного атома алкилкетонов.
Фталевая кислота – тривиальное название 1,2-бензолдикарбоновой кислоты. Ее получают окислением нафталина в жестких условиях.

В промышленности находит применение также каталитическое окисление орто-ксилола в аналогичных условиях.
Находящиеся по соседству карбоксильные группы придают фталевой кислоте некоторые свойства, нехарактерные для других дикарбоновых кислот. Так, в зависимости от условий, фталевая кислота образует два различающихся по структуре изомерных хлорангидрида.

Внутренний ангидрид фталевой кислоты, как и янтарный, в реакции с аммиаком дает циклический амид (фталимид), соединение которое используют для получения первичных алкиламинов (синтез Габриэля). Калиевая или натриевая соль фталимида, взаимодействуя с алкилгалогенидом или другим алкилирующим реагентом, превращается в соответствующий N-алкилфталимид. Последний вводят в реакцию с более мощным нуклеофилом – гидразином, в результате чего происходит переацилирование и выделяется первичный амин. Основным фактором, смещающим равновесие последней реакции вправо, является образование устойчивого ароматического гетероцикла – фталазин-1,4-диона (фталогидразида).

Для разложения N-алкилфталимида с целью получения алкиламина можно также применить гидролиз в кислой среде, но этот путь менее эффективен.
Источник
Способы получения дикарбоновых кислот
2.2. Дикарбоновые кислоты
Дикарбоновыми кислотами называют производные углеводородов, содержащие в своем составе две карбоксильные группы.
2.2.1. Алифатические дикарбоновые кислоты
По заместительной номенклатуре название дикарбоновых кислот строят исходя из соответствующих углеводородов с добавлением множительной приставки ди — и суффикса — овая кислота. Наряду с заместительной номенклатурой широко применяются тривиальные названия.

Для них характерна структурная изомерия.
2.2.1.1. Физические свойства
Дикарбоновые кислоты представляют собой кристаллические вещества. Низшие гомологи хорошо растворимы в воде. С увеличением молекулярной массы кислоты ее растворимость уменьшается.
Дикарбоновые кислоты получают теми же методами, что и моно-карбоновые кислоты, используя в качестве исходных веществ соответствующие бифункциональные соединения.
Окисление двупервичных гликилей, диальдегидов и гидроксикислот:

Одной из важных реакций получения дикарбоновых кислот является реакция омыления динитрилов:

2.2.1.2. Химические свойства
Имея в своем составе две карбоксильные группы, дикарбоновые кислоты диссоциируют ступенчато, образуя анион (рКа1) и дианион (рКа2).
Высокая кислотность по первой ступени объясняется взаимным влиянием второй карбоксильной группы, которая способствует делокализации образующегося отрицательного заряда карбоксилат-иона и тем самым повышает его устойчивость. По мере удаления карбоксильных групп ослабевает их взаимное влияние и кислотность по первой ступени падает. Отрыв протона от второй карбоксильной группы происходит труднее вследствие низкой стабильности дианиона, поэтому кислотность дикарбоновых кислот по второй ступени значительно ниже, чем по первой.
При максимальном удалении карбоксильных групп последние не оказывают взаимного влияния и через 5–6 связей каждая из них ведет себя независимо.
По химическим свойствам дикарбоновые кислоты, так же как и монокарбоновые, способны образовывать одни и те же функциональные производные. Только в зависимости оттого, одна или две карбоксильные группы участвуют в реакции, получают кислые или средние соли, полные и неполные эфиры, галогенангидриды, амиды и др. Например:

Вместе с тем следует отметить, что дикарбоновые кислоты проявляют и ряд специфических свойств. В частности, они по-разному относятся к нагреванию .
1. Отношение дикарбоновых кислот к нагреванию. Щавелевая и малоновая кислоты при нагревании свыше температуры плавления отщепляют оксид углерода (IV) и превращаются в монокарбоновые кислоты:

При нагревании янтарной и глутаровой кислот, взаимное влияние карбоксильных групп которых слабее, декарбоксилирования не происходит, а осуществляется процесс внутримолекулярной дегидратации с образованием циклических ангидридов:

Адипиновая кислота в этих условиях подвергается декарбоксилированию и дегидратации с образованием циклического кетона – циклопентанона:

2. Образование циклических амидов. При нагревании янтарной и глутаровой кислот или их ангидридов с аммиаком образуются циклические имиды:

2.2.2. Ароматические дикарбоновые кислоты
Ароматическими дикарбоновыми кислотами называют производные ароматических углеводородов, содержащие две карбоксильные группы, непосредственно связанные с ароматическим ядром.
Важными представителями этого класса являются фталевая, изофталевая и терефталевая кислоты:

Они имеют общее название – фталевые кислоты .
2.2.2.1. Способы получения
Основным способом получения ароматических дикарбоновых кислот является каталитическое окисление ксилолов кислородом воздуха. Так, при окислении п-ксилола получают терефталевую кислоту:

Фталевую кислоту получают из о-ксилола или нафталина:

2.2.2.2. Физические и химические свойства
Фталевые кислоты представляют собой кристаллические вещества с высокими температурами плавления. Фталевая и терефталевая кислоты мало растворимы в воде, в то время как изофталевая кислота в воде легко растворима.
По степени кислотности они превышают бензойную кислоту.
Арендикарбоновые кислоты образуют кислые и средние соли, полные и неполные сложные эфиры и амиды и т. д. Фталевая кислота в отличие от своих изомеров при нагревании легко теряет молекулу воды с образованием ангидрида, который при взаимодействии с аммиаком образует фталимид:

2.3. Функциональные производные карбоновых кислот
К важнейшим функциональным производным карбоновых кислот относятся: галогенангидриды, ангидриды, сложные эфиры, амиды, гидразиды, гидроксамовые кислоты, нитрилы и др.
2.3.1. Галогенангидриды карбоновых кислот (ацилгалогениды)
Ацилгалогениды – это такие производные карбоновых кислот, в которых гидроксильная группа, входящая в состав карбоксильной, замешена на атом галогена:

Названия ацилгалогенидов образуют из названий соответствующих кислот или ацильных групп и названий галогенов:

2.3.1.2. Способы получения
Хлор- и бромангидриды могут быть получены при действии галогенирующего реагента на карбоновые кислоты:

Йодпроизводные карбоновых кислот получают следующим образом:

2.3.1.3. Физические свойства
Низшие галогенангидриды карбоновых кислот – жидкости с резким запахом, раздражающие слизистые оболочки.
2.3.1.4. Химические свойства
Галогенангидриды – сильные электрофильные реагенты, сильнее карбоновых кислот. Галоген в этих соединениях обладает исключительно большой подвижностью.
Электрофильные свойства подобных соединений зависят от величины дробного положительного заряда ( δ +) на углероде карбонильной группы. Со стороны галогена проявляется выраженный –I, который в статическом состоянии больше + М , поэтому на атоме углерода возникает выраженный δ +, что и обуславливает сильные электрофильные свойства галогенангидридов. Они легко вступают в реакции нуклеофильного замещения:

В подобных реакциях в молекулу нуклеофила вводится ацильная группа, поэтому такие реакции называют реакциями аиилирования, а галогенангидриды карбоновых кислот – ацилирующими агентами.
Галогенангидриды в силу своей высокой активности нашли чрезвычайно большое применение в органическом синтезе.
2.3.2. Ангидриды карбоновых кислот
Ангидридами называют производные карбоновых кислот, в молекулах которых атом водорода карбоксильной группы замешен на ацильную группу:

Названия ангидридов образуют из тривиальных названий соответствующих кислот:

2.3.2.2. Способы получения
1. Дегидратация карбоновых кислот. При пропускании паров кислоты над соответствующим катализатором (пентаоксид фосфора, трифторуксусный ангидрид) происходит выделение воды – реакция дегидратации:

2. В промышленном масштабе ангидриды получают взаимодействием галогенангидридов с безводными солями карбоновых кислот:

3. Взаимодействие карбоновых кислот с кетенами (промышленный метод получения уксусного ангидрида):

2.3.2.3. Физические свойства
Ангидриды – кристаллические вещества или жидкости, малорастворимые в воде, обладают резким запахом.
2.3.2.4. Химические свойства
Ангидриды карбоновых кислот имеют менее выраженный электрофильный характер, чем галогенангидриды, но большую электрофильность, чем соответствующие карбоновые кислоты, поскольку +М- эффект атома кислорода приходится на две ацильные группы:

Ангидриды карбоновых кислот легко вступают в реакции нуклеофильного замещения, используются как ацилирующие реагенты:

Уксусный ангидрид используется в синтезе синтетических волокон, фармацевтических препаратов (ацетилсалициловая кислота).
2.3.3. Сложные эфиры
Сложные эфиры – производные карбоновых кислот, в молекулах которых гидроксильная группа, входящая в состав карбоксильной группы, замещена на остаток спирта или фенола –OR’:

Сложные эфиры называют по исходным кислоте и спирту или фенолу:

2.3.3.2. Способы получения
1. Взаимодействие галогенангидридов и ангидридов карбоновых кислот со спиртами и феноксидами щелочных металлов:

2. Взаимодействие карбоновых кислот со спиртами, катализируемое протоном кислоты (реакция этерификации):

Равновесие наступает, когда прореагирует примерно 2/3 моля исходного вещества. Для смещения равновесия в сторону образования конечных продуктов отгоняют полученный эфир или берут какое-либо из исходных веществ в избытке.
2.3.3.3. Физические свойства
Сложные эфиры – чаще жидкости с приятным запахом с более низкими температурами кипения, чем соответствующие кислоты и спирты, что связано с отсутствием ассоциации.
2.3.3.4. Химические свойства
Сложные эфиры – типичные электрофилы. Из-за +М-эффекта атома кислорода, связанного с углеводородным радикалом, они проявляют менее выраженный электрофильный характер по сравнению с галогенангидридами и ангидридами кислот:

Электрофильность эфиров увеличивается, если углеводородный радикал образует с атомом кислорода сопряженную систему, т. н. активированные эфиры:

Сложные эфиры вступают в реакции нуклеофильного замещения.
1. Гидролиз сложных эфиров проходит как в кислой, так и в щелочной среде.
Кислотный гидролиз сложных эфиров – последовательность обратимых превращений, противоположных реакции этерификации:

Механизм этой реакции включает протонирование атома кислорода карбонильной группы с образованием карбкатиона, который реагирует с молекулой воды:

Щелочной гидролиз. Гидролиз в присутствии водных растворов щелочей проходит легче, чем кислотный потому, что гидроксид-анион белее активный и менее объемный нуклеофил, чем вола. В отличие от кислотного, щелочной гидролиз необратим:

Щелочь выступает не в роли катализатора, а в роли реагента. Гидролиз начинается с нуклеофильной атаки гидроксид-ионом атома углерода карбонильной группы. Образуется промежуточный анион, который отщепляет алкоксид-ион и превращается в молекулу карбоновой кислоты. Алкоксид-ион, как более сильное основание, отрывает протон от молекулы кислоты и превращается в молекулу спирта:

Щелочной гидролиз необратим потому, что карбоксилат- анион имеет высокую делокализацию отрицательного заряда и не восприимчив к атаке спиртового гидроксила.
Часто щелочной гидролиз сложных эфиров называют омылением. Термин произошел от названия продуктов щелочного гидролиза жиров – мыла.
2. Взаимодействие с аммиаком (аммонолиз) и его производными протекает по механизму, аналогичному щелочному гидролизу:

3. Реакция переэтерификации (алкоголю сложных эфиров) катализируется как минеральными кислотами, так и щелочами:

Для смещения равновесия вправо отгоняют более летучий спирт.
4. Сложноэфирная конденсация Кляйзена характерна для эфиров карбоновых кислот, содержащих атомы водорода в α- положении. Реакция протекает в присутствии сильных оснований:

Если в реакцию вступают два сложных эфира, содержащие α- атомы водорода, то образуется смесь четырех возможных продуктов. Реакция используется для промышленного получения ацетоуксусного эфира.
5. Восстановление сложных эфиров:

Первичные спирты образуются при действии газообразного водорода в присутствии скелетного никелевого катализатора (никель Ренея).
Сложные эфиры имеют большое значение как ацилирующие реагенты, растворители, используются для синтеза альдегидов, кетонов, полимеров («органическое стекло» – плексиглас), лекарственных веществ: этилформиат – для производства витамина В2. Бензилбензоат используют для лечения чесотки. Эти вещества известны как отдушки в парфюмерии (этилформиат, этилацетат) и компоненты пищевых эссенций: грушевой – изоамилацетат, яблочной – изоамилвалерат, ромовой – этилформиат, ананасовой – этилбутират.
2.3.4. Амиды карбоновых кислот
Амидами называют производные карбоновых кислот, в молекулах которых гидроксильная группа карбоксила замещена на аминогруппу:

Эти соединения можно рассматривать как ацильные производные аммиака, первичных и вторичных аминов.
Названия амидов образуют от названий соответствующих кислот и аминов. В тривиальных названиях ацилов заменяют суффикс -ил на -амид. Согласно заместительной номенклатуре ИЮПАК в названиях соответствующих кислот часть -овая кислота заменяется на суффикс -амид. Символом N обозначают положение заместителей у атома азота амидной группы:

2.3.4.2. Способы получения
Амиды получают в результате взаимодействия галогенангидридов, ангидридов или сложных эфиров карбоновых кислот с аммиаком, первичными или вторичными аминами; при нагревании аммонийных солей карбоновых кислот, при гидролизе нитрилов:

2.3.4.3. Физические свойства
Амиды – кристаллические вещества или жидкости, растворимые в воде и органических растворителях. Это ассоциированные соединения, в которых π -электроны карбонильной группы смешаются к наиболее электроотрицательному атому кислорода, пара электронов азота смещена в сторону кислорода. За счет этого водород, находящийся при атоме азота, обладает способностью к образованию водородной связи с другой молекулой амида:

Поскольку амиды являются ассоциированными, то они имеют более высокие, по сравнению с соответствующими карбоновыми кислотами, температуры плавления и кипения.
2.3.4.4. Химические свойства
Амиды – очень слабые электрофилы.
Как же распределена электронная плотность в амидах?
За счет сильного смещения неподеленнои пары электронов атома азота (+M-эффект) к карбонильной группе частичный положительный заряд на атоме углерода С=O группы в амидах меньше, чем у галогенангидридов, ангидридов и сложных эфиров:

Вследствие такого электронного строения амиды практически не вступают в реакции с нуклеофильными реагентами.
1. Амфотерность амидов. Амиды – нейтральные вещества. Они проявляют амфотерный характер.
Основные свойства. Амиды можно рассматривать как производные аммиака, у которого атом водорода замещен на ацильныи остаток. Но ацильныи остаток содержит карбонильную группу, находящуюся в сопряжении с неподеленнои парой электронов атома азота, поэтому основные свойства NH2 группы значительно понижены. Амиды образуют соли лишь с сильными минеральными кислотами, вырывая пару электронов из р, π -сопряжения:

Солеобразование наступает в отсутствие влаги. Эти соли быстро разлагаются водой, так как они образуются слабым основанием и сильной кислотой.
Итак, по сравнению с аммиаком основные свойства амидов понижены вследствие сильного смещения пары электронов в сторону карбонильной группы:

Кислотные свойства. Однако по сравнению с аммиаком амиды обладают большей кислотностью. В молекулах незамещенных и N-замешенных амидов атомы водорода связи N–Н приобретают подвижность за счет сопряжения неподеленной пары электронов атома азота с л-электронами карбонильной группы.
Такие амиды проявляют свойства NH-кислот:

2. Гидролиз амидов. В нейтральной среде амиды гидролизуются значительно труднее, чем другие функциональные производные карбоновых кислот. Катализируют этот процесс кислоты или щелочи:

3. Дегидратация. При нагревании незамещенных амидов с сильными водоотнимаюшими средствами (Р2O5 или РOС13) образуются нитрилы:

4 . Восстановление амидов под действием алюмогидрида лития LiAlH4 идет до образования аминов:

N-замешенные амиды дают вторичные или третичные амины.
Полученные N-галогенамиды – нестабильные соединения со свойствами окислителя. Они используются в качестве галогенирующих реагентов.
Амиды карбоновых кислот находят широкое применение как растворители (формамид, диметилформамид и др.), в производстве синтетических волокон, лакокрасочных материалов, биологически активных веществ. Они довольно часто используются для идентификации кислот. Для подтверждения того, что получена та или иная кислота различными способами, можно ее идентифицировать по ее производным, в том числе амидам.
Например, четкая температура плавления амида масляной кислоты часто служит для однозначного решения вопроса о получении соответствующей кислоты, которая при нормальных условиях представляет собой жидкость.
2.3.5. Нитрилы (цианиды)
Нитрилы – органические соединения, содержащие одну или несколько цианогрупп –C=N, связанных с углеводородным радикалом:

Названия нитрилов образуют из тривиальных названий ацильных остатков соответствующих кислот или систематических названий карбоновых кислот, имеющих то же количество атомов углерода, с последующим добавлением суффикса — нитрил :

2.3.5.2. Способы получения
1. Дегидратация амидов сильными водоотнимаюшими средствами:

2. Замещение других групп цианогруппой – взаимодействие галогеналканов с солями циановодородной кислоты (цианидами):

2.3.5.3. Физические свойства
Нитрилы – жидкие или твердые вещества, не растворяющиеся в воде, но растворимые в органических растворителях нейтрального характера.
2.3.5.4. Химические свойства
Цианогруппа, проявляя отрицательный индуктивный эффект, увеличивает подвижность атомов водорода при α -углеродном атоме и за счет этого возможны реакции конденсации. Атомы углерода и азота образуют полярную связь, по месту разрыва которой нитрилы вступают в реакции нуклеофильного присоединения.

1. Присоединение нуклеофилъных реагентов. Гидролиз нитрилов проходит при нагревании с водными растворами кислот или щелочей с образованием амидов, которые гидролизуются до кислот:

Реакции со слабыми нуклеофилами обычно катализируются кислотами:
2. Реакция с участием α -углеродных атомов. Конденсация нитрилов (реакция Торпа) проходит в присутствии оснований: амидов, алкоксидов металлов. Реакция аналогична альдольной конденсации:

3. Восстановление нитрилов алюмогидридом лития или водородом протекает до соответствующих аминов:

Нитрилы широко используются в органическом синтезе. С помощью нитрила муравьиной кислоты в структуре соединения увеличивают углеродную цепь. Ацетонитрил – хороший растворитель для жирных кислот, используется в производстве витамина В12. Кристаллическое вещество – малонодинитрил легко вступает в реакции конденсации, широко используется для получения гетероциклических соединений: витаминов В6, В12, пестицидов, красителей.
Жиры (триацилглицерины, триацилглицериды) – сложные эфиры трехатомного спирта глицерина и высших алифатических кислот. Их общая формула:

В состав жиров преимущественно входят одноосновные кислоты с неразветвленнои цепью, которые содержат четное число углеродных атомов от 4 до 26.
По кислотному составу триаиилглицериды подразделяют на простые, содержащие остатки одинаковых кислот (R=R’=R») и смешанные, в составе которых встречаются разные кислотные остатки.
Природные жиры – чаше смешанные триацилглицерины. Кислотный состав жиров человеческого организма главным образом представлен стеариновой, пальмитиновой кислотами, которые поступают как с пишей, так и образуются путем биосинтеза. Ненасыщенные жирные кислоты: олеиновая, линолевая, арахидоновая не образуются в организме человека, а лишь поступают с пишей. Они получили название незаменимые.
По систематической номенклатуре ИЮПАК для жиров в начале перечисляют в алфавитном порядке названия жирных кислот, а затем указывают родоначальную структуру – глицерин. Согласно тривиальной номенклатуре в жирах часть названия жирных кислот — иновая заменяют суффиксом — ин :

Для синтеза триацилглицеринов можно использовать реакции O-ацилирования глицерина (этерификация, взаимодействие глицератов натрия с хлорангидридами кислот):

Синтетические способы получения жиров из глицерина не имеют промышленного значения. Чаще триацилглицерины выделяют из измельченных растительных и животных тканей экстракцией, прессованием, вытапливанием.
2.3.6.3. Физические свойства
Жиры – твердые или жидкие, но не газообразные вещества. Консистенция жиров зависит от их кислотного состава. Твердые триацилглицерины, как правило, содержат остатки насыщенных жирных кислот, это чаше жиры животного происхождения. В состав жидких жиров, которые называют маслами, входят в основном остатки ненасыщенных кислот. Растительные жиры, как правило, – жидкие. Исключение составляет масло какао, которое при нормальных условиях твердое вещество, и рыбий жир – жидкость.
Природные жиры являются смесями триацилглицеринов, поэтому не имеют четких температур плавления. Жиры нерастворимы в воде, но хорошо растворяются в органических растворителях: углеводородах, эфире, хлороформе.
2.3.6.4. Химические свойства
Как сложные эфиры жиры способны к гидролизу, а при наличии в их структуре ненасыщенных кислот триацилглицериды проявляют свойства алкенов.
1. Гидролиз жиров катализируют разбавленные растворы кислот или щелочей:

В промышленности гидролиз ведут при нагревании с водой в присутствии сульфокислот или нагреванием паром. Нередко для гидролиза используют фермент липазу.
При взаимодействии жиров с водными растворами щелочей образуется смесь глицерина и натриевых (калиевых) солей жирных кислот, которые называют мылами. Сам процесс щелочного гидролиза триацилглицеридов, ведущий к получению мыла, имеет название омыление. Этим же термином часто обозначают реакции щелочного гидролиза других соединений.
Полученную в результате гидролиза смесь пальмитиновой и стеариновой кислот используют для изготовления свечей.
Остановимся на качестве жиров. В аналитической практике реакция омыления жиров используется для установления их качества. Определяют так называемое число омыления – количество мг КОН, расходуемых на гидролиз 1 г жира. Избыток калия гидроксида нагревают с триацилглицеридом и методом обратного титрования определяют количество шелочи, которая ушла на нейтрализацию кислот. Таким образом определяют обшее содержание как свободных, так и связанных в триглицериды кислот.
2. Гидрогенизация жиров – присоединение водорода по месту разрыва двойных связей в остатках ненасыщенных кислот. Процесс проходит в присутствии никелевого или платинового катализатора при повышенной температуре и давлении:

Гидрогенизации подвергаются в основном растительные жиры и жиры морских животных. Этот процесс лежит в основе производства маргарина, мыла. Жидкие жиры омыляют, получают глицерин и непредельные кислоты, которые восстанавливают, а из них получают мыло.
Маргарин – пишевой жир с добавлением вкусовых веществ и отдушек, например, диацетила со вкусом и цветом сливочного масла. Промышленные жиры, полученные в результате гидрогенизации, называют саломасами.
3. Присоединение галогенов к жирам имеет большое аналитическое значение. Остатки ненасыщенных кислот в структуре жира обнаруживают по обесцвечиванию бромной воды.

4. Окисление жиров. Наличие двойных связей в молекулах жиров способствует легкой окисляемости, что ведет к «прогорканию». Различают два типа «прогоркания»: гидролитическое – расщепление до свободных кислоте короткой цепью, которое происходит под действием ферментов либо микроорганизмов, и окислительное. Окисление жиров ведет к образованию альдегидов и кетонов с короткой цепью, имеющих неприятный запах и вкус:

Жиры, образованные насыщенными жирными кислотами, при окислении образуют кетоны.
Источник