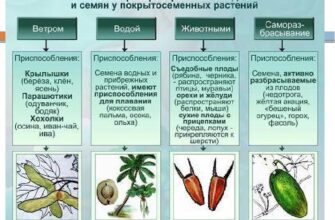
Портал аналитической химии
Методики, рекомендации, справочники
Метод равного помутнения (Гей-Люссака). Если титровать раствор NaBr раствором AgNOe (или наоборот), то происходит реакция:
,Очевидно, что осадок AgBr выпадает только до тех пор, пока в растворе еще имеется избыток Br-. Поэтому, отбирая в .конце титрования небольшие порции титруемого раствора и прибавляя к ним одну каплю разбавленного в 10 раз раствора AgN03, можно
точно уловить момент прекращения образования осадка. В данном случае это сильно облегчается потому, что вблизи точки эквивалентности осадок AgBr, коагулируя, собирается на дне сосуда в виде крупных творожистых хлопьев. Раствор при этом быстро осветляется, чему способствует энергичное перемешивание или встряхивание его.
При осаждении AgCl у которого произведение растворимости (1,78•1O-10) не так мало, как у AgBr (5,6•1O-13), методику работы приходится несколько усложнять. Дело в том, что насыщенный раствор AgCl, получающийся в точке эквивалентности, дает отчетливое помутнение как с раствором AgNO3, так и с раствором NaCl (понижение растворимости AgCl от введения одноименных ионов).
-ионов, то помутнение при добавлении AgNO3 будет, очевидно, большим, чем при добавлении NaCl. Наоборот, если раствор немного перетитрован, то NaCl вызовет более сильное помутнение, чем AgNO3.
Следовательно, для фиксирования точки эквивалентности здесь приходится перед концом титрования отбирать по две одинаковые пробы раствора и действовать на одну из них каплей раствора AgNO3, на другую — каплей раствора NaCl такой же концентрации. Титрование заканчивают при получении одинаковой интенсивности помутнения обеих проб.
«Метод равного помутнения», несмотря на то что приходится отбирать для проб некоторую часть титруемого раствора, является одним из точнейших методов титриметрического анализа. Однако он требует навыка и довольно кропотлив. Поэтому на практике обычно пользуются аргентометрическими методами с применением индикаторов.
Титрование до точки просветления. Метод титрования до точки просветления может быть применен тогда, когда малорастворимое соединение в процессе титрования находится в коллоидном состоянии. Например, при титровании I- раствором соли серебра, частицы AgI, адсорбируя I-, получают отрицательные заряды (см. § 27). Наличие зарядов, как известно, препятствует объединению частиц в более крупные агрегаты и оседанию их на дно сосуда. Вследствие этого при титровании сначала образуется не осадок, а коллоидный раствор AgI.
* Как будет показано позже, при рассмотрении титрования с внешними индикаторами ошибку, связанную с отбором проб, можно сделать исчезающе малой. Метод равного помутнения, предложенный в 1832 г. Гей-Люссаком, t явился одним из первых методов титриметрического анализа. Впоследствии он был использован для весьма точного определения атомных весов галогенов и серебра.
Источник
Портал аналитической химии
Методики, рекомендации, справочники
Метод равного помутнения (Гей-Люссака). Если титровать раствор NaBr раствором AgNOe (или наоборот), то происходит реакция:
,Очевидно, что осадок AgBr выпадает только до тех пор, пока в растворе еще имеется избыток Br-. Поэтому, отбирая в .конце титрования небольшие порции титруемого раствора и прибавляя к ним одну каплю разбавленного в 10 раз раствора AgN03, можно
точно уловить момент прекращения образования осадка. В данном случае это сильно облегчается потому, что вблизи точки эквивалентности осадок AgBr, коагулируя, собирается на дне сосуда в виде крупных творожистых хлопьев. Раствор при этом быстро осветляется, чему способствует энергичное перемешивание или встряхивание его.
При осаждении AgCl у которого произведение растворимости (1,78•1O-10) не так мало, как у AgBr (5,6•1O-13), методику работы приходится несколько усложнять. Дело в том, что насыщенный раствор AgCl, получающийся в точке эквивалентности, дает отчетливое помутнение как с раствором AgNO3, так и с раствором NaCl (понижение растворимости AgCl от введения одноименных ионов).
-ионов, то помутнение при добавлении AgNO3 будет, очевидно, большим, чем при добавлении NaCl. Наоборот, если раствор немного перетитрован, то NaCl вызовет более сильное помутнение, чем AgNO3.
Следовательно, для фиксирования точки эквивалентности здесь приходится перед концом титрования отбирать по две одинаковые пробы раствора и действовать на одну из них каплей раствора AgNO3, на другую — каплей раствора NaCl такой же концентрации. Титрование заканчивают при получении одинаковой интенсивности помутнения обеих проб.
«Метод равного помутнения», несмотря на то что приходится отбирать для проб некоторую часть титруемого раствора, является одним из точнейших методов титриметрического анализа. Однако он требует навыка и довольно кропотлив. Поэтому на практике обычно пользуются аргентометрическими методами с применением индикаторов.
Титрование до точки просветления. Метод титрования до точки просветления может быть применен тогда, когда малорастворимое соединение в процессе титрования находится в коллоидном состоянии. Например, при титровании I- раствором соли серебра, частицы AgI, адсорбируя I-, получают отрицательные заряды (см. § 27). Наличие зарядов, как известно, препятствует объединению частиц в более крупные агрегаты и оседанию их на дно сосуда. Вследствие этого при титровании сначала образуется не осадок, а коллоидный раствор AgI.
* Как будет показано позже, при рассмотрении титрования с внешними индикаторами ошибку, связанную с отбором проб, можно сделать исчезающе малой. Метод равного помутнения, предложенный в 1832 г. Гей-Люссаком, t явился одним из первых методов титриметрического анализа. Впоследствии он был использован для весьма точного определения атомных весов галогенов и серебра.
По мере того как все больше и больше I- связывается Ag+, частицы AgI постепенно теряют адсорбированные ими 1_-ионы, и заряд их уменьшается. В конце концов заряд уменьшается настолько, что происходит коагуляция частиц и осаждение их в виде крупных творожистых хлопьев. Раствор при этом совершенно осветляется. Этот момент, называемый точкой просветления, в некоторой степени зависит от степени разбавления раствора иодида и от интенсивности перемешивания раствора при титровании.
При большом разбавлении раствора KI и сильном перемешивании точка просветления практически совпадает с точкой эквивалентности. Для количественного определения 1—ионов (или Ag+) раствор KI разбавляют приблизительно до концентрации 0,004 н. (например, к 20 мл
0,1 н. раствора прибавляют
500 мл воды) и титруют 0,1 н. раствором AgNO3 (прибавляя его небольшими порциями при энергичном перемешивании) до точки просветления.
Понятно, что в титруемом растворе не должно быть двух- или многозарядных катионов, так как они вызвали бы преждевременную коагуляцию золя AgI. Точно так же можно титровать до точки просветления раствор соли свинца молибдатом аммония (NH4)2Mo04 (при этом образуется осадок PbMoO4).
Методы с применением индикаторов
Наиболее часто при аргентометрическом титровании пользуются в качестве индикаторов растворами хромата калия K2CrO4 (в методе Мора) или железо-аммонийных квасцов NH4Fe(SO4J2 (в методе Фольга рда).
Применение K2CrO4 в качестве индикатора основано на способности CrO4- давать с Ag+ кирпично-красного цвета осадок Ag2CrO4, который в определенных условиях начинает выпадать лишь после того, как определяемые С1
-ионы будут практически полностью осаждены в виде AgCl.
Причина этого заключается в различии величин растворимости хлорида и хромата серебра.
Поэтому и осаждаться должен первым именно AgCl. Поскольку, однако, произведение [Ag+][Cl-] остается все время (приблизительно) постоянным, по мере осаждения Cl- в виде AgCl концентрация Ag+ в растворе должна постепенно повышаться *. При этом в конце концов окажется достигнутой и та концентрация Ag+-HOHOB, которая необходима для того, чтобы началось осаждение Ag2CrO4, 1,05- Ю-5 г-ион/л.
С этого момента наряду с AgCl начнет осаждаться также и Ag2CrO4, и взмученный в жидкости осадок приобретает красновато-бурую окраску, при получении которой заканчивают титрование.
Таким образом, в указанных условиях выпадение осадка Ag2CrO4 действительно начинается только после практически полного осаждения С1—ионов в виде AgCl.
Найденной выше концентрации остающихся в растворе С1—ионов отвечает величина рС1 =—Ig 1,05-10-6 « 5,03, лежащая внутри области скачка на кривой титрования (4—6). Это свидетельствует о. том, что данный индикатор при концентрации его
Ю-2 M дает возможность достаточно точно фиксировать точку эквивалентности при титровании.
Метод Мора применяют для определения серебра, хлоридов и бромидов (определять иодиды и роданиды этим методом нельзя, так как результаты сильно искажаются вследствие явлений адсорбции).
Что бы ни определялось по методу Мора — соли галогенов или соли серебра, порядок титрования должен быть всегда такой же, как при установлении титра раствора AgNO3. Другими словами, всегда нужно к измеренному объему раствора соли галогена приливать раствор соли серебра из бюретки, так как только в этом . случае получается резкое изменение окраски в конце титрования.
Нужно, далее, иметь в виду, что метод Мора применим только для титрования в нейтральной или слабощелочной среде (рН 6,5—10), так как Ag2CrO4 растворим в кислотах и в их присутствии не выпадает *.
Если анализируемый раствор имеет кислую реакцию, его нейтрализуют раствором тетрабората натрия Na2B4O7-IOH2O или бикарбоната натрия NaHCO3. Эти растворы не должны содержать примесей хлоридов, в чем необходимо убедиться, подействовав на небольшое количество раствора, подкисленного HNO3, AgNO3.
Другим условием применимости метода Мора является отсутствие в исследуемом растворе катионов, дающих с Сг04- осадки. Таковы, например, Ba2+, Pb2+, Bi3+ и др. Мешают также некоторые анионы, образующие осадки с Ag+ (например, PO4-, AsO4-, C2Q4-и т. п.). Все это сильно ограничивает применимость рассматриваемого метода. Более широкое применение имеет роданометрический метод (метод Фольгарда, роданометрия).
Это дает возможность титровать растворы солей серебра стандартным раствором NH4SCN (или KSCN) в присутствии индикатора— раствора соли железа (III), например железо-аммонийных квасцов NH4Fe(SO4)Z-12H2O.
* В сильнощелочной среде (рН > 10) образуется осадок Ag2O. В присутствии солей аммония диапазон рН должен быть сужен до 6,5—7,2, иначе выделяющийся аммиак будет связывать ионы серебра в комплекс [Ag(NH3)2]+. Понятно, что метод Мора неприменим также в присутствии солей, имеющих вследствие гидролиза кислую реакцию.
Пока не достигнута точка эквивалентности, концентрация остающихся в растворе SCN
hohob настолько мала, что образования комплексов железа, в частности Fe(SCN)2+, не происходит. Но первая же избыточная капля раствора NH4SCN повысит эту концентрацию настолько, что указанная реакция произойдет и раствор приобретет более или менее интенсивную оранжево-красную окраску.
Применяя способ обратного титрования (титрование по остатку), этот метод можно использовать также и для определения бромидов и хлоридов. Например, роданометрическое определение бромидов проводится по схеме:
Br- + Ag+ (избыток) —>•AgBr + Ag+ (остаток)
Ag+ (остаток) + SCN- —► AgSCN (титрование)
Так же определяются и хлориды.
Конец титрования можно сделать более отчетливым, прибавляя к титруемому раствору 1—2 мл нитробензола C6H5NO2, четыреххлористого углерода CCl4 или хлороформа CHCl3. Эти вещества, адсорбируясь на поверхности осадка AgCl, сильно замедляют реакцию между ним и роданидными комплексами железа.
Еще лучше к исследуемому раствору хлорида в мерной колбе прилить измеренный избыток стандартного раствора AgNO3 и разбавить водой до метки. После этого часть раствора фильтруют через сухой фильтр и измеренный пипеткой объем фильтрата титруют раствором роданида. В этом случае осадок AgCl оказывается отделенным от раствора и помешать титрованию не может.
На практике в качестве индикатора применяют насыщенный раствор железо-аммонийных квасцов NH4Fe(SO4J2• 12H2O с небольшим количеством концентрированной HNO3 для подавления гидролиза, вследствие которого раствор приобретает бурую окраску.
В отличие от метода Мора в этом методе присутствие кислоты не только не вредит титрованию, но, наоборот, способствует получению более точных результатов.
Источник
Способы фиксирования момента эквивалентности
В ряде случаев окислительно-восстановительного титрования точки эквивалентности фиксируют по изменению окраски титруемого раствора, вызываемой избытком окрашенного стандартного раствора (например, перманганата).
В методах, основанных на титровании стандартным раствором иода, или на выделении иода (иодометрия), точку эквивалентности устанавливают при помощи индикатора — крахмала, специфически реагирующего с иодом (в присутствии свободного иода бесцветный раствор крахмала окрашивается в интенсивно синий цвет; в присутствии избытка восстановителей, восстанавливающих нейтральный иод, интенсивно синее окрашивание исчезает).
В некоторых случаях конец титрования определяется по обесцвечиванию кроваво-красной окраски роданида железа в присутствии избытка восстановителя (титанометрия).
Перечисленные вещества можно отнести к группе специфических реактивов (индикаторов), действующих в пределах определенных методов окисления—восстановления.
Помимо этой группы веществ (индикаторов), в методах окисления—восстановления широко применяются индикаторы, называемые ред-окс-индикаторами, которые изменяют окраску в зависимости от величины окислительно-восстановительного потенциала (Е).
Иногда для более четкого фиксирования точки эквивалентности прямое визуальное титрование проводят либо в неводных растворах, либо к водному раствору добавляют небольшое количество органического растворителя, не смешивающегося с водой (бензол, изоамиловый эфир, четыреххлористый углерод и т. п.). При этом органический растворитель экстрагирует слабоокрашенное вещество, и окраска его усиливается. Так, например, поступают при иодометрическом титровании, экстрагируя иод.
В ряде случаев используют внешние, флюоресцирующие, хемилюми-несцентные и другие индикаторы (см. ниже).
Наиболее широко для фиксирования конечной точки титрования применяют физико-химические (инструментальные) методы (потенциометрический, амперометрический, кулонометрический, кондуктометрический, хронокондуктометрический, высокочастотный, фотометрический, спектрофотометрический и др.).
Источник
Точка эквивалентности и способы ее фиксации
Точка эквивалентности (Т. Э.) — такая точка титрования, в котором количество прибавленного титранта Т эквивалентно количеству титруемого вещества.
Наступающий в процессе титрования момент, в котором количество раствора реагента (р.в.) становится теоретически строго эквивалентным количеству определяемого компонента (о.в.), реагирующего с прибавляемым р.в. согласно определенному уравнению химической реакции, называют моментом (точкой) эквивалентности (т.э.).
Момент, при котором заканчивают титрование, называют конечной точкой титрования (к.т.т).
А конечную точку титрования мы определяем при помощи индикаторов, которые изменяют свою окраску — это и будет подтверждение точки эквивалентности.
Методы с применением индикаторов:
Наиболее часто при аргентометрическом титровании пользуются в качестве индикаторов растворами
1)хромата калия K2CrO4 (в методе Мора)
Применение K2CrO4 в качестве индикатора основано на способности CrO4- давать с Ag+ кирпично-красного цвета осадок Ag2CrO4
2)железо-аммонийных квасцов NH4Fe(SO4J2 (в методе Фольга рда).Пока не достигнута точка эквивалентности, концентрация остающихся в растворе SCN
hohob настолько мала, что образования комплексов железа, в частности Fe(SCN)2+, не происходит. Но первая же избыточная капля раствора NH4SCN повысит эту концентрацию настолько, что указанная реакция произойдет и раствор приобретет более или менее интенсивную оранжево-красную окраску.Применяя способ обратного титрования (титрование по остатку), этот метод можно использовать также и для определения бромидов и хлоридов.
Гей-Люссака Безиндикаторный Титрование до просветления, т.е. когда новая порция титранта не вызывает помутнения раствора Br — Мора K2Cr2O7 Появление в щелочной среде кирпичного цвета осадка Ag2CrO4 при избытке титранта Cl Br — Фольгарта FeCl3 Появление красного окрашивания при избытке раствора NH4CNS в качестве титранта Ag+ (прямое титрование);
34. Индикатор — средство, позволяющее путем визуального наблюдения установить достижение системой определенного состояния ( определенной концентрации анализируемого вещества).
Основные типы индикаторов
Известные в настоящее время визуальные индикаторы делят на несколько типов по очень важным в практическом отношении признакам: 1) химической природе, типу реакции взаимодействия индикатора с соответствующим веществом титриметрической системы; 2) характеру обратимости взаимодействия; 3) характеру окраски разных форм индикатора; 4) наблюдаемому эффекту; 5) способу применения.
В соответствии с типом реакции взаимодействия выделяют индикаторы присоединения или отдачи ионов и электронов.
Все индикаторы присоединения ионов имеют сходные признаки. Они отдают или присоединяют какую-либо частицу. Например, Н30 + или ОН», если это кислотно-основные (рН-индикаторы); Ме я+ — металлохромные (Me — металл; металло-индикаторы); Ме л+ или An w _ — осадкообразующие, адсорбционные индикаторы (Ап — анион). Индикаторы этой группы в разной мере, но обязательно взаимодействуют с ионом гидроксония, даже если не предназначены для его определения. По этой причине при определении ионов металлов необходимо поддерживать почти постоянной концентрацию ионов Н30 + , используя избыток сильной кислоты, основания или добавляя буферные смеси. Индикаторы присоединения электронов принимают участие в реакциях окисления-восстановления, и поэтому их называют окислительно-восстановительными или редоксиндикаторами. Их показания часто зависят как от концентрации фиксируемых частиц, так и от кислотности титруемого раствора. Их окраска изменяется замедленно, а химические реакции с их участием протекают с образованием промежуточных соединений.
По характеру обратимости индикаторной реакции индикаторы делят на обратимые и необратимые.
Обратимыми являются те индикаторы, для которых наблюдаемые изменения около точки перехода окраски происходят любое число раз в одну или другую сторону при изменении концентрации соответствующего компонента титриметрической системы. Так, фенолфталеин окрашивает раствор в розовый цвет при добавлении щелочи и обесцвечивается при добавлении избытка кислоты. Этот процесс может быть повторен практически неограниченно. Большинство кислотно-основных и металлохромных индикаторов относится к обратимым. Эти индикаторы удобны, потому что при случайном введении избытка титранта в локальную область титруемого вещества определение все же будет правильным; перемешивание приводит к выравниванию средней концентрации раствора, исчезновению местного оттитровывания и восстановлению окраски индикатора до первоначальной. Необратимыми являются индикаторы, для которых изменения вблизи точки перехода можно наблюдать только в одном направлении вследствие необратимых химических превращений, коренным образом изменяющих их структуру и реакционную способность. Индикаторами указанного типа являются многие типичные кислотно-основные индикаторы, относящиеся к азосоединениям и суль- фофталеинам, если их использовать при редоксиметрических титрованиях. Свойство индикаторов этого типа необратимо изменять свою окраску при локальном возрастании концентрации титранта, особенно при плохом перемешивании раствора, может быть причиной ошибочного фиксирования степени оттитрованности раствора.
По способу применения индикаторы делятся на внутренние и внешние. Индикатор, который вводят непосредственно в анализируемый раствор, называют внутренним. В некоторых случаях индикатор нельзя ввести в анализируемый раствор. Тогда отбирают одну-две капли этого раствора и помещают на специальную бумагу, пропитанную индикатором, или смешивают с раствором индикатора на белой фарфоровой пластинке или на обычной фильтровальной бумаге. Использованный индикатор является внешним по отношению к анализируемому раствору.
В соответствии с природой наблюдаемого эффекта различают индикаторы цвета — цветные; свечения — хемилюминесцентные и флуоресцентные; индикаторы образования гетерофазы (в частном случае — осадка) и изменения ее свойств — осадкообразующие, помутнения и адсорбционные. Из указанных индикаторов наиболее часто используют цветные индикаторы.
Цветные индикаторы бывают одно- и двухцветные. У одноцветных индикаторов окрашена только одна форма (вторая — бесцветна). Типичными представителями одноцветных индикаторов являются кислотно-основные индикаторы типа нитрофенолов разных степеней замещенности, а также фталеины (например, на- фтолфталеин, фенолфталеин, тимолфталеин).
Нитрофенол и его таутомерно-ионные формы:
В щелочной среде отщепление протона от группы —ОН бесцветной формы приводит к появлению хиноидной группировки в одном из бензольных колец и появлению окраски. Фенолфталеин:
У двухцветных индикаторов окрашены обе равновесные формы. Большинство цветных индикаторов принадлежит именно к этому типу, независимо от вещества, на изменение концентрации которого индикатор реагирует. Как видно из приведенных примеров, причиной изменения окраски индикаторов является исчезновение одних и появление других функциональных группировок при переходе от одной формы к другой. Например, для метилового оранжевого наблюдается переход бензоидной группировки в хиноидную, а при увеличении кислотности раствора — азогруппы N-»N—- в группу ««N—NH—.
Метиловый оранжевый и его таутомерно-ионные формы:
Двухцветные индикаторы отличаются от одноцветных тем, что их основные характеристики (интервал перехода окраски и показатель титрования) в достаточно широких пределах не зависят от концентрации в растворе. В случае одноцветных кислотно-основных индикаторов десятикратное изменение их концентрации приводит к смещению интервала изменения окраски примерно на единицу pH.
Особое место среди цветных индикаторов занимают смешанные индикаторы, представляющие смесь индивидуальных индикаторов или какого-либо индикатора с инертным красителем. Формы компонентов смешанных индикаторов имеют взаимодополняющие цвета, при наложении которых раствор приобретает нейтральный серо-белый цвет, существующий в очень узком интервале концентраций определяемого компонента. Некоторые характеристики индивидуальных красителей и их смесей приведены в табл. 6.2. Смешанные индикаторы отличает четкое изменение окраски, т.е. контрастный переход. Это изменение происходит в узком интервале концентраций определяемого вещества. С этими индикаторами удобно работать в условиях искусственного освещения, в слабо- окрашенных и мутных растворах. Смешанные индикаторы значительно расширяют круг веществ, которые можно определять тит
| Состав индикатора | ^pH|nd ИЛИ pTCMtuliKIU | Окраска форм индикатора (кислотная — шелочная) |
| I. Метиловый красный | 4,2-6,2 | Красная — желтая |
| II. Метиленовый голубой | Инертный краситель | Синяя —синяя |
| III. Смесь I и II (1: 1) | 5,4 (серая) | Красно-фиолетовая— зеленая |
| IV. Бромкрезоловый зеленый | I ОС rn | Желтая—синяя |
| V. Смесь I и IV(1: 3) | 5,1 (серая) | Винно-красная — зеленая |
рованием. К таким относятся, прежде всего, вещества, для которых характерна малая величина константы равновесия реакции титрования, что определяет малую величину скачка титрования (например, 0,2—0,6 ед. pH или рМе).
В некоторых случаях целесообразно использовать специфические индикаторы. Они реагируют только со строго определенным веществом с четко выраженным аналитическим эффектом. Например, в иодометрии роль специфического индикатора выполняет крахмал. Он образует с иодом аддукт, имеющий синию окраску. Специфический индикатор на Fe 3+ — роданид-ион, NCS». При их взаимодействии образуется комплексный ион FeNCS 2+ , имеющий карминово-красный цвет. Индикаторов этого типа известно немного, но их поиск проводится постоянно.
Достижения системой определенного состояния при титровании можно зафиксировать, используя самоиндикацию, т.е. появление или исчезновение окраски раствора вследствие появления или исчезновения какого-либо компонента титриметрической системы — титранта или титруемого вещества. Так, при титровании перманганатом калия появление его в растворе окрашивает последний в розовый цвет. Наоборот, если КМп04 титруется соответствующим восстановителем, то при оттитоовывании его первоначальная розовая окраска раствора исчезает. Аналогичная картина наблюдается при использовании в качестве титранта растворов солей Ce(IV). Указанный принцип фиксирования конца титрования широко используют при титриметрических определениях органических веществ.
Основные характеристики индикаторов. Индикаторы принято характеризовать такими параметрами, как интервал (область) перехода эффекта (окраски, свечения, помутнения раствора) и показатель титрования.
Под интервалом перехода (окраски, свечения, помутнения раствора) понимают интервал концентраций (или рХ) фиксируемого вещества, в пределах которого происходит заметное для глаза изменение аналитического эффекта. Этот интервал обычно обозначают как ApX|nd. Значение рХ, при котором наиболее резко заметно изменение аналитического эффекта, называют показателем титрования ( точкой перехода) индикатора и обозначают как pT)nd.
Взаимосвязь величины интервала перехода и показателя титрования можно установить, используя уравнение константы равновесия индикаторной реакции XInd + Ind». Найдя р-функ- цию равновесной концентрации фиксируемой частицы, получают:
За начало и конец интервала перехода принимают значения концентрации X, при которых наблюдается эффект, характерный только для какой-то одной формы индикатора, т.е. вначале свободной — Ind», а затем связанной — XInd. В случае цветных индикаторов условно считают, что окраска раствора определяется формой «Ind
», если соотношение [Ind»]: [XInd] = (6. 10): 1 и, наоборот, формой «XInd», если это соотношение равно 1 :(6. 10). Поэтому интервал перехода имеет величину ApX|nd.
Интервалы перехода и показатели титрования цветных индикаторов
| Метод титрования | X | К\п& | рТы | ApX|nd |
| Протолитометрия | н + ОН” | Ка/К5Кь/К5 | Pallid) pA*(Ind) | рКа<Ь)(Ш)± (0,8-1) |
| Комплексометрия | Ме л+ | p'(Melnd) | pp'(Melnd) | pp'(MeInd)± (0,8-1) |
| Редоксиметрия | Е | A£indJc//0,059 | AEjnd | A^ind ± 0,059 |
В действительности, значение pTind смещено от середины в сторону значения рХ, соответствующего форме с более интенсивным аналитическим эффектом, например окраской. Это смещение может составлять 0,2—0,4 единицы рХ.
Интервал перехода индикатора и его показатель титрования зависят от природы индикатора (^^(Ind)’, p'(Melnd), Ai?in 4 . 10′ 5 ) М, что соответствует добавлению одной—трех капель 0,5—0,1 %-ного раствора индикатора на каждые 10—25 мл титруемого раствора. Количество титранта, взятое на титрование каждой аликвоты (пипетки) анализируемого раствора, должно быть одним и тем же, особенно в случае одноцветных индикаторов. Введение меньшего,
чем указано, количества индикатора делает раствор слабоокра- шенным, а большего — излишне окрашенным. Наблюдать изменение окраски таких растворов (особенно в конце титрования) трудно, часто даже невозможно.
Источник