Измерение потенциала проводника
Как мы уже отмечали ранее, поле внутри проводника имеет нулевую напряженность. Следовательно, он является эквипотенциальным по всему объему. Иными словами, значения потенциалов будут одинаковы во всех его точках.
Разность потенциалов двух любых точек проводника будет равна:
Потенциал проводника – это значение его потенциала, одинаковое для всех точек.
Рассмотрим ситуацию с изолированным заряженным проводником. Вокруг него имеется электрическое поле, создаваемое зарядом и распространяемое в веществе вокруг него. Нормировка потенциала будет равна нулю в бесконечности. Тогда его потенциал может быть выражен так:
Интегрирование может начинаться в любой точке проводника и заканчиваться в бесконечности.
Измерение с помощью электрометра
Электроскоп — прибор для измерения разности потенциалов между двумя проводниками.
Если его стрелка или листочки заключены в металлическую оболочку, то его называют электрометром. Для измерения нам надо соединить один проводник с его оболочкой, а второй – с шариком, после чего стрелка прибора примет потенциал измеряемого тела. При этом образуется электрическое поле с силовыми линиями, направленными от стрелки к оболочке или наоборот. От напряженности и конфигурации этого поля будет зависеть величина отклонения стрелки. Важно отметить, что поле внутри металлической оболочки не будет зависеть от внешнего поля, а будет определяться только разностью потенциалов между стрелкой и оболочкой.
Мерой разности потенциалов двух измеряемых тел является угол отклонения стрелки электрометра.
Градуировка на таком приборе может быть и в вольтах. Зачастую при измерении вторым телом выступает земля, то есть выполняется заземление оболочки электрометра. В таком случае его показания будут означать потенциал тела относительно Земли.
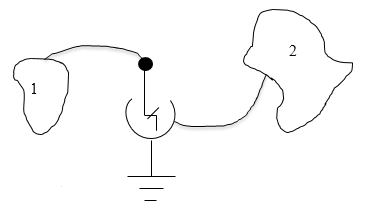
Можно заземлять как оболочку, так и шарик, это не имеет значения. Это определит только направление, в котором будут идти силовые линии, а угол отклонения стрелки окажется одинаковым.
Очевидно, что стрелка должна иметь слабую связь с внешними полями, чтобы точность измерения электрометром была высокой. Однако слишком сильная связь искажает показания. Чтобы создать нужный уровень защиты, в оболочке экрана или шарика, а также в наружной части стержня, соединяющего стрелку с шариком, оставляют небольшое отверстие. Если контакт с внешними полями будет слишком интенсивным, то на этих частях прибора возникнут посторонние заряды, индуцированные внешними полями, которые будут вносить искажения при переходе на стрелку. По той же самой причине провода, соединяющие измеряемые тела, не должны быть толстыми.
С помощью электрометра мы можем убедиться в эквипотенциальности поверхности проводника. Соединив прибор с разными точками заряженного проводника, мы увидим, что отклонение стрелки останется прежним.
Измерение с помощью метода электрического зонда
Если нам нужно измерить разность потенциалов в жидких или газообразных диэлектриках, то применяется метод электрического зонда. Это небольшой металлический прибор, состоящий из шарика или диска, соединенного проволокой с шариком электрометра. При этом прибор должен иметь заземленную оболочку.
Зонд необходимо поместить в нужную точку диэлектрика, после чего он покажет разность потенциалов между оболочкой и стрелкой (или между зондом и Землей). Нужно учитывать, что помещение зонда в диэлектрик сильно изменяет потенциал измеряемой точки. Это происходит из-за индукционных зарядов на шарике прибора и самом зонде. Чтобы получить достоверные данные, нужно, чтобы при внесении зонда прибор и шарик электроскопа приняли исходный потенциал измеряемой точки.
Убрать индукционные заряды можно разными способами.
Например, если зонд капельный, то нам потребуется небольшой сосуд с проводящей жидкостью, на дне которого есть маленькое отверстие. Через него капли проводника унесут индукционный заряд, и все заряды с противоположным знаком перейдут на стрелку электрометра. Это должно изменить угол отклонения стрелки.
Если зонд не заряжен, то его потенциал такой же, как у окружающего его пространства. Поскольку он соединяется с шариком электрометра, то его потенциал будет равен ему. В итоге мы получим нужное значение потенциала без искажений.
Также индукционные заряды удаляют при помощи так называемого пламенного зонда. В таком случае в качестве зонда выступает конец металлической проволоки, соединенный с диэлектрической трубкой, используемой в качестве газовой горелки.
Высокая температура слегка ионизирует воздух вокруг и делает его проводящим. В итоге индукционные заряды уносятся ионами вместе с потоком газа. Примерно та же идея лежит в основе радиоактивного зонда.
Примеры решения задач
Условие: экспериментально подтверждена отрицательная заряженность Земли. Около земной поверхности имеется напряженность, среднее значение которой составляет примерно 130 В на кв.м. У человека имеется разность потенциалов между головой и ногами, равная примерно 200 В . Поясните, почему при этом не происходит поражения электрическим током.
Решение
Тело человека – очень хороший проводник, значит, оно вносит сильные искажения в электрическое поле вокруг себя. На поверхности человеческого тела заряды перераспределяются, но это продолжается весьма недолгое время, и интенсивность процесса невысока. Положение силовых линий поля по отношению к телу является перпендикулярным, а эквипотенциальные поверхности огибают его точно так же, как металлический предмет. Все тело человека является эквипотенциальным, т.е. в разных его точках потенциалы одинаковы. Напряженность поля зависит от разности потенциалов поля, если разность потенциалов равна нулю, значит и напряженность поля будет нулевой.
Ответ: Именно поэтому человек не чувствует разности потенциалов электрического поля Земли.
Условие: прикосновение к электроскопу пальцем вызывает его разрядку. Будет ли прибор разряжаться в том случае, если рядом с ним будет находиться заряженное тело, изолированное от Земли?
Решение
При поднесении заряженного тела к электроскопу мы увидим, что на стержне прибора появятся индуцированные заряды. Со стороны внешнего конца они будут иметь знак, противоположный зарядам на внутреннем конце.
Ответ: электроскоп не разрядится.
Условие: измерение с помощью электрического зонда показало, что потенциал электрического поля Земли меняется по мере подъема вверх примерно на 100 В / м . Подсчитайте, чему будет равен заряд Земли, если считать, что поле создается именно им. Радиус Земли считать равным 6400 к м .
Решение
То, что модуль напряженности меняется, может быть связано с изменением потенциала Земли. Нам потребуется формула:
Учитывая размерность, сделаем вывод, что в задаче нужно использовать именно E .
Зная теорему Остроградского-Гаусса, можем записать:
Здесь S = 4 π R 2 , где поверхность, через которую рассмотрен поток вектора напряженности. Она совпадает со сферой радиуса Земли.
Искомый заряд выражается так:
Примем ε = 1 . Подставим это в формулу, учтем, что S = 4 π R 2 , и получим:
q = ∆ φ ∆ x 4 πR 2 εε 0 .
Переведем радиус Земли в С И , получим: R = 6 , 4 · 10 6 м . Вычислим заряд Земли:
q = 100 · 4 · 3 , 14 · 8 , 85 · 10 — 12 · 6 , 4 · 10 6 2 1 ≈ 4 , 55 · 10 5 К л
Ответ: Земля имеет заряд, равный 4 , 55 · 10 5 К л .
Источник
Методы измерения потенциала.
Существует два метода измерения потенциала:
 |
Метод прямого измерения потенциала основан на том, что к входу измерительного прибора подключается пара электродов. Схема прямого измерения потенциала представлена на рисунке 17.
Естественно, что измерительный прибор весьма и весьма сложен. Во-первых, он должен иметь высокое внутреннее сопротивление. Чтобы более строго подойти к решению этого вопроса, необходимо логически рассуждать: общий потенциал параэлектрода равен сумме потенциалов индикаторного электрода и электрода сравнения:
Дальше потенциал будет распределяться внутри самого электрода и часть падать на прибор. Все это можно расписать через силу тока, которая в данной цепи одинакова, через сопротивление прибора и внутреннее сопротивление электрода:
Е общ. = IR внутр + IR прибора
Для того, чтобы наиболее точно измерить потенциал, падение потенциала на приборе должно быть много больше, чем внутри электрода. Из этого вытекает следующее условие:
Это самая упрощенная схема. В самом деле, в измерительной технике сотня радиодеталей: конденсаторы, сопротивления, усилители, трансформаторы. Это устройство намного сложнее, чем приемник. Но чтобы понять принцип, мы нарисовали так. Здесь имеется реостат, ручку которого нужно двигать до состояния, когда гальванометр покажет нуль. Когда гальванометр покажет нуль, то вольтметр будет показывать соответствующую разность потенциалов, напряжение между точками А и В. Компенсационный метод является идеальным с точки зрения точного измерения потенциала, т.к. отсутствует систематическая ошибка, связанная с потерей потенциала на внутреннем параэлектроде. В последнее время выпускают прямопоказывающие приборы для работы с ионоселективными электродами. Они выпускаются двух типов: это С-анализаторы и рС-анализаторы. Во втором случае прибор показывает lgС. Как следует из уравнения Нернста потенциал пропорционален рС, а также lgС, и поэтому шкала прибора градуирована в единицах рС. Довольно простой случай — это К-анализаторы. В электродную схему прибора включают антилогарифматор, т.е. С – это антилогарифм для потенциала. Прибор измеряет потенциал, значит нужно антилогарифмировать. Сразу должен отметить, что строгой техники, которая позволяет точно рассчитать антилогарифм, в электронике нет. Это должны быть специальные ЭВМ. В простых приборах отсутствует антилогарифматор, поэтому используются различные приближенные варианты. Наибольшее значение в практике приобрели прямопоказывающие С-анализаторы. Это анализаторы на калий, натрий, хлориды и нитраты. Один из анализаторов, нитратомер, нами был выпущен для Беларуси большой партией около 10000 штук. В настоящее время вы его часто сможете встретить на практикумах.
Количественные методы в потенциометрии
Для определения количественного содержания вещества потенциометрию можно применять в двух вариантах.
В первом варианте аналитическим сигналом является потенциал пары электродов (индикаторного и сравнения) в соответствующем растворе. Измерив значение потенциала, можно определить содержание вещества при помощи градуировочного графика либо по уравнению Нернста.
Во втором варианте потенциометрического титрования аналитическим сигналом является объем титранта, а ионометрия используется для индикации точки эквивалентности. Оба метода широко применимы.
Одно из важнейших достоинств потенциометрии – высокая экспрессность, и в этом смысле она наиболее идеальный вариант..
Вернемся к первому методу – прямой потенциометрии
Метод базируется на Нернстовской функции электрода. Аналитический сигнал в ионометрии – потенциал, обычно имеет не очень простую зависимость от lgС. В зависимости от величины и знака заряда Нернстовская функция электрода может иметь вид:

Рис.19. Определение рабочей области функционирования электрода
Согласно уравнению Нернста зависимость Е – lg a линейна во всем интервале активностей. В незабуференных системах эта зависимость носит более сложный характер.
По этой зависимости можно выделить три участка. На первом участке потенциал описывается уравнением Нернста. Область активностей или концентраций определяемых ионов, соответствующих этому участку является рабочей областью ИСЭ.
На втором участке потенциал ИСЭ практически не меняется с изменением активности определяемых ионов в растворе. Этот участок определяет предел обнаружения электрода, или ту минимальную концентрацию (активность) ионов, которую с точностью «да» или «нет» может уловить ИСЭ. Для большинства ИСЭ предел обнаружения составляет 10 -4 –10 -8 м. Приблизительно эта концентрация соответствует точке пересечения асимптот к линиям участков I и II. Более строго эта концентрация соответствует точке, в которой отклонение зависимости Е – lg a от асимптоты к участку I составит:
Характерно, что в забуференных сопряженных системах второй участок не наблюдается.
Третий участок характеризуется также потерей чувствительности ИСЭ к определяемому иону. Одной из причин появления этого участка, который в случае электродов на основе нейтральных переносчиков имеет еще более странный вид, объясняется экстракцией соли определяемого катиона в фазу пленочной или жидкой мембраны с каким либо противоионом.
 |
Нернстовский участок имеет наклон в зависимости от величины и знака заряда ионов.
 |
 |
 |  |
Для однозарядных анионов q = –58 мВ (рис.21), для однозарядного катионов q = 58 мВ (рис.23), для 2-зарядных анионов q =- 29мВ (рис. 22) и для 3-зарядных катионов q = 19 мВ ( рис.24). Таким образом, очевидно, что, получив электродную функцию и построив зависимость по уравнению Нернста, всегда можно определить знак заряда и величину заряда потенциалопределяющего иона. Глядя на график для анионов, можно сказать, что он не строго описывается уравнением Нернста. Он должен иметь вид прямой линии. В области высоких концентраций, а иногда и в области низких концентраций практически всегда наблюдается потеря чувствительности к определяемому иону и отклонение от нернстовской зависимости. Появление этих отклонений для цели анализа нежелательно, т.к. приводит к снижению чувствительности электрода, т.е. снижению или увеличению предела обнаружения. Малые концентрации, а их наиболее важно измерять, в данном случае измеряться не могут.
 |
Рис. 25. Определение рабочей области функционирования электрода
Три линии – асимптоты к линейным участкам.
Согласно рекомендациям ИЮПАК, точки пересечения этих линий определяют нижний и верхний пределы обнаружения ионоселективного электрода. Имея такой график и найдя точки пересечения, мы сможем найти пределы обнаружения, то есть те концентрации, ниже или выше которых электрод не является, строго говоря, прибором для проведения анализа.
Математическая интерпретация предела обнаружения следующая: в этой точке уровень шумов или уровень фона в отсутствие определяемых ионов и уровень вклада в сигнал потенциалопределяющих ионов равны, т.е. аналитический сигнал удваивается за счет сложения фона и полезного сигнала.. Если принять это условие, то нетрудно подсчитать, что в этой точке реальная зависимость отличается от идеальной на qlg2.
В очень большом числе методик берется такой подход: полезный сигнал равен фону (равен шуму), и эта точка является предельной точкой для обнаружения данного иона. Не всегда такое условие выдвигается, но очень часто.
Прежде чем остановиться на причинах появления нижнего предела обнаружения, нужно отметить, что аналогичная ситуация возникает весьма часто и в области высоких концентраций. Как в области низких концентраций, так и в области высоких концентраций применим тот же закон: точка пересечения является точкой верхнего предела обнаружения.
Пределы обнаружения могут находиться как графически, так и расчетно. Расчетно ПО находятся как точки, в которых экспериментальные кривые отклоняются от теоретических на qlg2. Из пределов обнаружения наиболее важной характеристикой является нижний предел обнаружения. Это связано с тем, что разбавить пробу значительно проще, чем сконцентрировать.
Есть различные варианты концентрирования определяемых ионов. Самым простым является упаривание анализируемого раствора, либо экстракционное концентрирование из большого объема пробы малым объемом экстрагента.
Концентрирование можно провести также ионообменно, сорбционно, дистилляцией и т.д. В области высоких концентраций нужно только разбавить в нужное количество раз фоном. А низкие концентрации сконцентрировать намного сложнее и не всегда возможно. Поэтому нижние пределы обнаружения электродов обычно входят в паспортные данные наряду с коэффициентами их селективности.
Перед вами данные по нижнему пределу обнаружения целого ряда ионов.
| Анионы | НПО, М |
  ClO4 — Au(CN)2 — F — HgI3 — I — ClO4 — Au(CN)2 — F — HgI3 — I — |      |
| Катионы | НПО, м |
| K + | |
| NH4 + | |
| Ca 2+ | |
| Na + | |
| Ag + | |
| Hg +2 |  |
Причины появления НПО
Для целого ряда электродов причиной появления нижнего предела обнаружения является растворение электродноактивного вещества в анализируемом растворе. Идеально это наблюдается в случае осадочного хлорид-селективного электрода на основе AgCl/.
 M. НПО = 1×10 -5 М.
M. НПО = 1×10 -5 М.
Для AgBr уже наблюдается расхождение – экспериментальный НПО оказывается более высоким, чем рассчитанный. То же наблюдается и для электрода на основе AgI.
В случае жидкостных электродов наблюдается другая ситуация: растворимость электродноактивного вещества в подавляющем большинстве случаев близка к нулю или находится на уровне 10 -10 -10 -15 М. Например, тетрадециламмония нитрат. Для жидкостных и пленочных электродов рассчитанная растворимость электродноактивного вещества оказывается намного ниже, чем реальные пределы обнаружения. Рассчитанные 10 -10 -10 -11 М, а экспериментальные гораздо выше. Одной из основных причин этого является асимметричный выход определяемых ионов в анализируемый раствор за счет протекания ионообменной реакции. Попробуем это объяснить подробнее.
Симметричный выход отмечается при растворении осадка. В этом случае концентрация аниона равна концентрации катиона. В случае тетрадециламмония нитрата катион никогда не перейдет в водную фазу, он слишком гидрофобный. Но в анализируемом растворе присутствуют другие ионы. Даже в дистиллированной воде это Н + , ОН — и НСО3 — , который появляется из воздуха. В стеклянной же посуде появляются ионы за счет растворения стекла. Вытеснение определяемых ионов ионами примесей по ионообменному механизму и обуславливает появление более высоких значений пределов обнаружения. В данном случае они могут быть описаны следующим образом:

где В – примесный ион, А – определяемый ион,  — концентрация определяемого иона в мембране.
— концентрация определяемого иона в мембране.
Это уравнение взято из константы обмена. Концентрация определяемых ионов будет равна Ö из произведения константы обмена на концентрацию определяемых ионов в мембране и умноженных на концентрацию анионов примеси в водном растворе. Это уравнение можно легко вывести самим. Оказывается, что по этой схеме можно полуколичественно предсказать нижний предел обнаружения в реальных средах.
Нернстовская функция часто нарушается и по другим причинам. Наиболее часто встречающиеся отклонения представлены на рисунке 26.

 |
В данном случае мы видим нернстовскую функцию К-селективного электрода для различных солей калия. Почти прямая линия — это для КCl. Это явление называют появление анионных функций К-селективного электрода. Причина этого связана с экстракцией калиевых солей в фазу мембраны валиномицином. Валиномицин экстрагирует калий в мембрану в виде соответствующей калиевой соли. Реакция экстракции протекает по следующему уравнению:
Концентрация продукта будет пропорциональна концентрации калиевой соли KClO4 в квадрате. Это приводит к тому, что концентрация калия в мембране растет даже быстрее, чем в воде, т.к. появляется квадратичная зависимость. Мы помним, что основой стабильности любого электрода является стабильная постоянная концентрация определяемого иона в мембране, она не должна меняться. Если она меняется, то будет меняться и потенциал. Но, учитывая, что в уравнении есть квадрат, то это не просто потеря чувствительности, а даже обратная анионная функция, но она замаскирована.
Очень часто, в особенности для нитратного электрода наблюдается такой тип отклонения от нернстовской функции. В области разбавленных растворов с уменьшением концентрации определяемого иона наблюдается скачкообразное возрастание потенциала.
Появление такого отклонения обусловлено тем, что в мембране электрода накапливается электродноактивное вещество в форме более гидрофильных анионов, если перед измерением электрод долгое время контактировал с раствором, содержащим высокие концентрации примесных гидрофильных анионов. Это не случайная ситуация, она встречается всегда. Дело в том, что нитраты определяют на фоне алюмокалиевых квасцов примерно 1М концентрации, т.е. в растворе очень много сульфат-ионов. Если электрод долгое время контактировал с этим раствором, где мало нитратов, а много квасцов, то сульфат в значительной мере вытесняет нитрат-ион, особенно на поверхности электрода из мембраны и занимает его место. Далее, когда берем малую концентрацию нитрат-иона, наблюдается обратный процесс: нитраты входят в мембрану, и когда их очень мало, раствор фактически обедняется нитратами и создается иллюзия, что наблюдаются отклонения. В самом же деле электрод показывает строго, но только в приэлектродном слое, а не вообще в растворе. Зависимость носит характер кривой титрования, это фактически и есть титрование нитратами в сульфатной зоне ЧАС в мембране. Этот скачок наблюдается при равенстве концентраций нитратов в растворе и сульфатной формы ЧАС в мембране.
Второй случай отклонений – снижение нернстовских наклонов градуировочной функции. В критических случаях дело доходит до появления половинных наклонов. Появление половинных наклонов наблюдается в схожем, но обратном случае при накоплении в мембране значительно более гидрофобных ионов, чем определяемый ион.
 |
Например, в случае нитратного электрода это будут анионы алкилсульфатов, перхлората, бората и ряда других. В этом случае мембрана электрода окажется полностью в форме более гидрофобного иона, т.е. электрод эксплуатируется там, где много мешающих гидрофобных ионов, и потихоньку они накапливаются в мембране в такой мере, что наблюдаются отклонения. Это тоже ситуация реальная, а не гипотетическая. Этот случай тоже описывается указанным уравнением.
По этой причине для всех электродов крайне редки теоретические наклоны. Всегда для любого электрода можно рассчитать теоретический наклон: заряд известен, t 0 и все остальные величины постоянны, но на практике наклон электрода редко когда бывает теоретическим. Например, для F-электрода q = 55-56 мВ, а не 58-59, и это хороший результат. Если q ниже 50 мВ, то тогда прибор не градуируется, а дает информацию о смене электрода. Все эти причины приводят к тому, что при высокой экспрессности метод прямой потенциометрии обладает невысокой точностью. В идеальных условиях, если потенциал мерить специальной техникой с высокой точностью, если температуру стабилизировать, вводить специальные добавки, то точность составляет 2-3 относительных %.
Первой причиной ошибок является неточность измерения потенциала измерительной техникой. Рассмотрим таблицу, в которой приведены относительные ошибки в % определения для ионов с зарядом 1, 2 и 3 при ошибке в потенциале 0.1, 0.5, 1, 2, 2.5 мВ. Трехзарядные ионы – это редкий случай. Особенно важный столбик для однозарядных ионов. Эти данные легко можно рассчитать из уравнению Нернста.
| % мв | Z =1, % | Z=2,% | Z=3, % |
| 0,1 | 0,4 | 0,8 | 1,2 |
| 0,5 | 2,0 | 4,0 | 6,0 |
| 1,0 | 4,0 | 8,0 | 12,0 |
| 2,0 | 8,0 | 16,0 | 24,0 |
| 2,5 | 10,0 | 20,0 | 30,0 |
Обычная измерительная аппаратура дает неточность 0.5 – 1мВ, прецизионная техника — 0.1мв. Ниже измерять точнее не имеет смысла, т.к. начнут доминировать другие ошибки.
Второй причиной является температурный и концентрационный дрейф индикаторных электродов и электродов сравнения – постоянное изменение потенциала в ту или иную сторону (+ 5%). Температурный дрейф вызван тем, что в стакане температура +10 0 , а в комнате +20 0 , т.е раствор нужно термостатировать перед измерением хотя бы с точностью ±1 0 .
Концентрационный дрейф – более сложная, но более понятная причина. Дело в том, что все электроды являются неравновесными системами. Если электрод включить в цепь и оставить на 1000 лет, то разности потенциалов всех электродов будут равны нулю, концентрации растворов изменятся так, что потенциалы уравняются. Элементы этого дрейфа всегда проявляются в зависимости от концентрации ионов в мембране, от ее проницаемости. Все электроды в бесконечности – это квазиравновесные системы. В данный момент все строго описывается, а проходят десятки лет, и все изменяется. Это причина может внести ±5 % в общую погрешность.
Третья причина еще более серьезная — это эффект ионной силы раствора. Если мы отградуировали электроды на фоне дистиллированной воды, а измеряем на фоне 0.1М раствора электролита, то обеспечено 20 % ошибки для однозарядных ионов, для 2-зарядных ионов – 40 % ошибки, для более высоких концентраций ошибка будет увеличиваться. Обязательный или почти обязательный прием при относительно точных измерениях – это разбавление фоном градуировочных растворов и анализируемого раствора. Для стабилизации ионной силы раствора применяют мировой стандартный буферный раствор БРОИС.
Еще одна причина – действие рН. Для стабилизации рН применяют, например, ацетатный буферный раствор, для определения нитратов – 1М фоновый раствор алюмокалиевых квасцов, т.е. в каждом конкретном случае можно придумать фоновый электролит, относительно инертный к определяемому электроду, и в то же время создающий нужную ионную силу. Естественно, что эффект ионной силы до конца устранить невозможно, понятно почему. Мы ведь не знаем, что есть в определяемом растворе, т.е. изначально присутствует какой-то фон, который не всегда известен, поэтому ионной силой мы только улучшаем ситуацию, но до конца устранить ее невозможно. Эти причины дают небольшие ошибки.
Грубые ошибки в измерении концентрации могут быть вызваны в том случае, если определяемый ион легко гидролизующийся и неправильно выбрано рН анализируемого раствора. Даже для фторида этот эффект уже проявляется. HF сильнее уксусной, и рН ниже 3 нежелательно, и связано это не столько с подавлением диссоциации HF, сколько с димеризацией, т.е. очень силен процесс образования димера, реакции гомосопряжения (образуется HF2 — ). Для фторид-аниона это не так страшно, гораздо страшнее для других анионов. Например, СО3 2— анион существует только при рН=14 (на 99% в этой форме). На более простые ионы, например, цианид-ион влияет диссоциация. Его определяют очень часто, между тем эта кислота имеет константу 10 -9 , и поэтому если даже работать при нейтральном рН=7, то ошибка в измерениях будет в 100 раз.
Последний случай – действие комплексообразования. Например, при определении F — возникает задача избавиться от комплексов F — c железом. Они очень прочные, и нужен демаскирующий агент, и туда вносят ацетат-анион (ацетатный буферный раствор). Гораздо сложнее с определением катионов 2- и 3-зарядных металлов. Эти катионы почти во всех средах находятся в форме каких-нибудь комплексов. Например, при рН >2 – гидроксокомплексы. Чтобы избавиться от них, надо подкислить среду, а чем? Ртуть, например, образует нитратные комплексы, особо прочные хлоридные комплексы, и многие другие, т.е очень трудно подобрать среду, в которой тяжелые металлы с большой молекулярной массой комплексообразования находились бы в форме катионов. Выход такой: наоборот, перевести их в форму какого-либо комплекса, например, HgI3 — и определять в форме комплекса (золото – в форме цианида, цинк – тетрароданоцинката и др.).
Рассмотрим методы, позволяющие существенно снизить ошибки в методе прямой потенциометрии.
Источник