Производство сульфата аммония
Сульфат аммония (NH4)2SО4 — бесцветное кристаллическое вещество, содержит 21,21 % азота. Сульфат аммония применяют почти исключительно в качестве удобрения; он обладает весьма небольшой гигроскопичностью, мало слеживается, внесение его в почву не вызывает затруднений. Недостатками являются низкое содержание азота и большая физиологическая кислотность. При его применении в почве, если она не содержит достаточного количества оснований, постепенно накапливается серная кислота, для нейтрализации которой необходимо периодически производить известкование.
Физико-химические основы получения сульфата аммония
Промышленные способы получения сульфата аммония в основном базируются на нейтрализации серной кислоты аммиаком. Для этой цели используют аммиак, содержащийся в газе, получаемом при коксовании каменных углей. Очистка коксового газа от аммиака (и одновременно от пиридиновых оснований) совмещается с производством сульфата аммония. Синтетический аммиак перерабатывают в более концентрированные азотные удобрения, например в нитрат аммония или в карбамид.
Нейтрализация серной кислоты газообразным аммиаком по реакции:
сопровождается выделением большого количества теплоты. Эта теплота (в сатураторном процессе) и теплота, подводимая извне (в бессатураторном процессе), расходуется на испарение из системы значительного количества воды и продукт кристаллизуется из пересыщенного раствора. Важно обеспечить кристаллизацию из горячего реакционного раствора средней соли, не допуская выделения кислых солей.
Находящиеся в серной кислоте примеси, особенно сульфаты железа и алюминия, затрудняют кристаллизацию сульфата аммония. При нейтрализации кислоты осаждаются коллоидные гидроксиды железа и алюминия:
обволакивающие кристаллы сульфата аммония и тормозящие их рост. Во избежание этого кислоту нейтрализуют не полностью — в непрерывно действующих реакторах поддерживают кислую реакцию среды.

Участки кривых соответствуют насыщению раствора: 1 – (NH4)2SО4;
2 – 4(NH4)2SО4∙Н2SО4; 3 – 3(NH4)2SО4∙Н2SО4; 4 – (NH4)2SО4 ∙ Н2SО4;
5 – (NH4)2SО4 ∙ 3Н2SО4
Рисунок 1 – Изотермы растворимости в системе (NH4)2SО4—Н2SО4—Н2О при температурах 10, 30, 50, 70 и 90 °С
В этой системе в твердой фазе могут существовать различные кислые соли. Поле кристаллизации сульфата аммония (NH4)2SО4лежит в области составов систем, содержащих небольшие количества серной кислоты – а1Е1с при температуре 10 °С и а2Е2с при температуре 90 °С. Во избежание выделения кислых солей содержание серной кислоты в жидкой фазе системы должно быть меньше, чем в точках Е, т. е. меньше 11,08 % при температуре 10 °С или 19,77 % при температуре 90 °С. В процессе нейтрализации реакционная масса имеет высокую температуру, но при последующем отделении кристаллов она охлаждается и это необходимо учитывать при выборе состава реакционного раствора. Практически кислотность раствора поддерживают на уровне 4-12 % свободной серной кислоты, распределяя серную кислоту в значительном количестве циркулирующего реакционного раствора.
Технологическая схема производства сульфата аммония
Основным сырьевым источником в производстве сульфата аммония является аммиак коксового газа. В коксовом газе содержится 6-14 г/м 3 аммиака. Его можно переработать в сульфат аммония тремя способами: косвенным, прямым и полупрямым.
По косвенному способу коксовый газ охлаждают, при этом из него конденсируется смола и надсмольная вода, насыщенная аммиаком; оставшийся в газе аммиак поглощают водой в аммиачных скрубберах. Из полученной аммиачной воды и из надсмольной воды отгоняют аммиак в дистилляционных колоннах, который затем поглощают серной кислотой. Этот способ требует громоздкого оборудования и значительного расхода энергии.
По прямому способу поглощение аммиака серной кислотой с образованием сульфата аммония производят непосредственно из коксового газа, предварительно охлажденного до температуры 68 °С и очищенного от смолы в электрофильтрах.
Наиболее экономичен и широко распространен полупрямой способ. Коксовый газ для конденсации смолы сначала охлаждают до температуры 25-30 °С. Конденсат расслаивается на два слоя: нижний — смолу и верхний — надсмольную воду, в которой растворена часть аммиака. Надсмольную воду обрабатывают в дистилляционной колонне известковым молоком и выделившийся аммиак поглощают серной кислотой вместе с аммиаком, оставшимся в доочищенном в электрофильтрах от смолы коксовом газе.
Поглощение аммиака из коксового газа можно производить в сатураторах барботажного типа (сатураторный способ) или в скрубберах (бессатураторный способ). В сатураторном способе поглощение аммиака из коксового газа и кристаллизация сульфата аммония совмещены в одном аппарате — сатураторе. Это ограничивает возможность выбора технологического режима, который был бы оптимальным одновременно для обоих процессов, т. е. обеспечивающего наиболее полное поглощение аммиака и образование крупнокристаллического сульфата аммония, легко отделяемого и отмываемого от маточного раствора. В бессатураторных способах, используемых на некоторых заводах, эти процессы ведут раздельно — поглощение аммиака в скрубберах, а кристаллизацию сульфата аммония — в кристаллизаторах.

Схема производства сульфата аммония сатураторным способом
Коксовый газ, охлажденный до температуры 25-30 °С и очищенный от смолы, поступает в подогреватель 1, где нагревается глухим паром до температуры 60-80 °С. Подогретый газ смешивается с аммиаком, полученным при переработке надсмольной воды, и направляется по барботажной трубе 5 в сатуратор 4.
Газ барботирует через 78 %-ный раствор серной кислоты, при этом образуется сульфат аммония:
В сатураторе одновременно с образованием сульфата аммония из газа извлекаются пиридиновые основания, образующие с серной кислотой комплексные соединения. Они разлагаются при температуре выше 65 о С с выделением пиридина, который удаляется из сатуратора вместе с газом. Тепло, необходимое для испарения избыточной влаги из образовавшегося раствора сульфата аммония, подводится в сатуратор с коксовым газом, подогретым в аппарате 1.
По выходе из сатуратора газ направляется в ловушку 2 для отделения от брызг кислоты, затем охлаждается и передается на дальнейшее использование. Когда кислотность раствора в сатураторе снижается до 6-8 % (что соответствует содержанию в нем 140-170 г/л связанного аммиака), из раствора начинают выделяться кристаллы сульфата аммония. Образующаяся пульпа центробежным насосом перекачивается в кристаллоприемник 8. Маточный раствор из верхней части кристаллоприемника переливается в приемный сосуд 6 и возвращается в сатуратор. Кристаллы сульфата аммония непрерывно поступают из кристаллоприемника в центрифугу 7, где отделяются от маточного раствора. Отфугованные кристаллы сульфата аммония, имеющие влажность около 2 %, передают на склад или направляют па сушку.
Часть раствора непрерывно циркулирует между сатуратором и баком 3. Благодаря циркуляции и непрерывному перекачиванию пульпы из сатуратора в кристаллоприемник с возвратом маточного раствора в сатуратор в нем обеспечивается постоянный.уровень жидкости и ее тщательное перемешивание. Поэтому кристаллы соли все время находятся во взвешенном состоянии, и рост кристаллов происходит равномерно во всей массе раствора.
Содержание свободной серной кислоты в маточном растворе, циркулирующем в сатураторе, должно быть в пределах 6-8 %. При понижении кислотности (до 1-2 %) из раствора выпадают более крупные кристаллы, что может вызвать забивку сатуратора солью; при этом также ухудшается поглощение аммиака из газа. С повышением кислотности раствора увеличивается растворимость в нем сульфата аммония и получаются более мелкие кристаллы. Если в растворе содержится более 11 % кислоты, образуется легкорастворимый в воде бисульфат аммония NH4HSО4.
На получение 1 т сульфата аммония затрачивают: 0,73-0,75 т серной кислоты (100 %-й), 0,26-0,27 т аммиака (содержащегося в 30-35 тыс.м 3 коксового газа), 100-108 МДж электроэнергии, 8 м 3 воды и 2,7-6 т пара.
К недостаткам сатураторного способа, помимо малого размера получаемых кристаллов, сильно пылящих при сушке, относится и большой расход энергии на преодоление гидравлического сопротивления абсорберов. Этих недостатков лишены бессатураторные способы.
В бессатураторных процессах абсорбцию аммиака из коксового газа ведут в полых скрубберах или кислым ненасыщенным раствором сульфата аммония с последующей вакуум-выпаркой на кристалл, или кислым насыщенным раствором с выращиванием образовавшихся мелких кристаллов в кристаллизаторах под атмосферным давлением. Чаще используют первый способ — орошение абсорбера ненасыщенным раствором предотвращает их засоление, а кристаллизация в выпарных аппаратах позволяет регулировать размеры получаемых кристаллов. Схема такого процесса показана на рисунке.

Схема производства сульфата аммония бессатураторным способом с вакуум-выпаркой
Аммиак улавливается в полом скруббере 2, снабженном форсунками. Скруббер разделен на две ступени. Нижняя его часть орошается раствором, содержащим 3-4 % свободной Н2SО4, верхняя — раствором, содержащим 10 — 12 % Н2SО4. Коксовый газ из скруббера проходит через ловушку брызг 1 и направляется на дальнейшую переработку. Серная кислота и вода (необходимая для разбавления и компенсации испарения) поступают в сборник 4 раствора, циркулирующего через верхнюю часть скруббера с помощью насоса 5. Часть этого раствора через смолоотделитель 3 подается в сборник 11 маточного раствора, циркулирующего через нижнюю часть скруббера с помощью насоса 12. Сюда же поступает маточный раствор с центрифуги 8.
Из нижней зоны скруббера часть раствора, в котором содержится около 1 % свободной серной кислоты и 40 % сульфата аммония, отбирается через смолоотделитель 3 в сборник 10 и насосом 9 подается в вакуум-выпарной аппарат 6. Образовавшиеся здесь кристаллы опускаются в нижнюю коническую часть аппарата, выполняющую роль кристаллорастителя, где мелкие кристаллы поддерживаются во взвешенном состоянии в восходящем потоке свежего раствора. Это обеспечивает их рост при небольшом пересыщении раствора, и более 60 % кристаллов сульфата аммония получаются с размерами, превышающими 0,5 мм. Такие же результаты достигаются при использовании выпарных аппаратов, снабженных выносными кристаллорастителями. Суспензия из выпарного аппарата, содержащая 50-60 % кристаллов, подается для фильтрования на центрифугу 8, где кристаллы промываются горячим конденсатом при температуре 70-80 °С для удаления остатков кислоты. Затем продукт направляется на сушку.
Источник
Производство сульфата аммония
Сульфат аммония (NH4)2SО4 — бесцветное кристаллическое вещество, содержит 21,21 % азота. Сульфат аммония применяют почти исключительно в качестве удобрения; он обладает весьма небольшой гигроскопичностью, мало слеживается, внесение его в почву не вызывает затруднений. Недостатками являются низкое содержание азота и большая физиологическая кислотность. При его применении в почве, если она не содержит достаточного количества оснований, постепенно накапливается серная кислота, для нейтрализации которой необходимо периодически производить известкование.
4.3.1 Физико-химические основы получения сульфата аммония
Промышленные способы получения сульфата аммония в основном базируются на нейтрализации серной кислоты аммиаком. Для этой цели используют аммиак, содержащийся в газе, получаемом при коксовании каменных углей. Очистка коксового газа от аммиака (и одновременно от пиридиновых оснований) совмещается с производством сульфата аммония. Синтетический аммиак перерабатывают в более концентрированные азотные удобрения, например в нитрат аммония или в карбамид.
Нейтрализация серной кислоты газообразным аммиаком по реакции:
сопровождается выделением большого количества теплоты. Эта теплота (в сатураторном процессе) и теплота, подводимая извне (в бессатураторном процессе), расходуется на испарение из системы значительного количества воды и продукт кристаллизуется из пересыщенного раствора. Важно обеспечить кристаллизацию из горячего реакционного раствора средней соли, не допуская выделения кислых солей.
Находящиеся в серной кислоте примеси, особенно сульфаты железа и алюминия, затрудняют кристаллизацию сульфата аммония. При нейтрализации кислоты осаждаются коллоидные гидроксиды железа и алюминия:
обволакивающие кристаллы сульфата аммония и тормозящие их рост. Во избежание этого кислоту нейтрализуют не полностью — в непрерывно действующих реакторах поддерживают кислую реакцию среды.
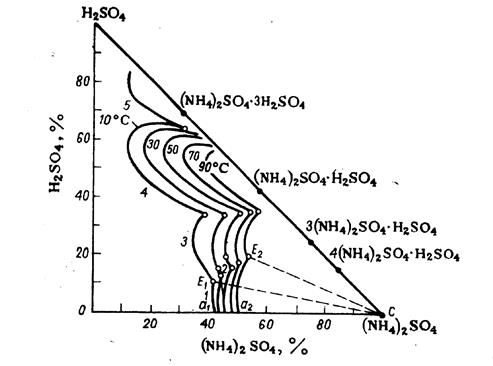
Участки кривых соответствуют насыщению раствора: 1 – (NH4)2SО4;
Рисунок 4.6 – Изотермы растворимости в системе (NH4)2SО4—Н2SО4—Н2О при температурах 10, 30, 50, 70 и 90 °С
В этой системе в твердой фазе могут существовать различные кислые соли. Поле кристаллизации сульфата аммония (NH4)2SО4 лежит в области составов систем, содержащих небольшие количества серной кислоты – а1Е1с при температуре 10 °С и а2Е2с при температуре 90 °С. Во избежание выделения кислых солей содержание серной кислоты в жидкой фазе системы должно быть меньше, чем в точках Е, т. е. меньше 11,08 % при температуре 10 °С или 19,77 % при температуре 90 °С. В процессе нейтрализации реакционная масса имеет высокую температуру, но при последующем отделении кристаллов она охлаждается и это необходимо учитывать при выборе состава реакционного раствора. Практически кислотность раствора поддерживают на уровне 4-12 % свободной серной кислоты, распределяя серную кислоту в значительном количестве циркулирующего реакционного раствора.
4.3.2 Технологическая схема производства сульфата аммония
Основным сырьевым источником в производстве сульфата аммония является аммиак коксового газа. В коксовом газе содержится 6-14 г/м 3 аммиака. Его можно переработать в сульфат аммония тремя способами: косвенным, прямым и полупрямым.
По косвенному способу коксовый газ охлаждают, при этом из него конденсируется смола и надсмольная вода, насыщенная аммиаком; оставшийся в газе аммиак поглощают водой в аммиачных скрубберах. Из полученной аммиачной воды и из надсмольной воды отгоняют аммиак в дистилляционных колоннах, который затем поглощают серной кислотой. Этот способ требует громоздкого оборудования и значительного расхода энергии.
По прямому способу поглощение аммиака серной кислотой с образованием сульфата аммония производят непосредственно из коксового газа, предварительно охлажденного до температуры 68 °С и очищенного от смолы в электрофильтрах.
Наиболее экономичен и широко распространен полупрямой способ. Коксовый газ для конденсации смолы сначала охлаждают до температуры 25-30 °С. Конденсат расслаивается на два слоя: нижний — смолу и верхний — надсмольную воду, в которой растворена часть аммиака. Надсмольную воду обрабатывают в дистилляционной колонне известковым молоком и выделившийся аммиак поглощают серной кислотой вместе с аммиаком, оставшимся в доочищенном в электрофильтрах от смолы коксовом газе.
Поглощение аммиака из коксового газа можно производить в сатураторах барботажного типа (сатураторный способ) или в скрубберах (бессатураторный способ). В сатураторном способе поглощение аммиака из коксового газа и кристаллизация сульфата аммония совмещены в одном аппарате — сатураторе. Это ограничивает возможность выбора технологического режима, который был бы оптимальным одновременно для обоих процессов, т. е. обеспечивающего наиболее полное поглощение аммиака и образование крупнокристаллического сульфата аммония, легко отделяемого и отмываемого от маточного раствора. В бессатураторных способах, используемых на некоторых заводах, эти процессы ведут раздельно — поглощение аммиака в скрубберах, а кристаллизацию сульфата аммония — в кристаллизаторах.

На рисунке 4.7 изображена схема производства сульфата аммония сатураторным способом.

Рисунок 4.7 – Схема производства сульфата аммония сатураторным способом
Коксовый газ, охлажденный до температуры 25-30 °С и очищенный от смолы, поступает в подогреватель 1, где нагревается глухим паром до температуры 60-80 °С. Подогретый газ смешивается с аммиаком, полученным при переработке надсмольной воды, и направляется по барботажной трубе 5 в сатуратор 4.
Газ барботирует через 78 %-ный раствор серной кислоты, при этом образуется сульфат аммония:
В сатураторе одновременно с образованием сульфата аммония из газа извлекаются пиридиновые основания, образующие с серной кислотой комплексные соединения. Они разлагаются при температуре выше 65 о С с выделением пиридина, который удаляется из сатуратора вместе с газом. Тепло, необходимое для испарения избыточной влаги из образовавшегося раствора сульфата аммония, подводится в сатуратор с коксовым газом, подогретым в аппарате 1.
По выходе из сатуратора газ направляется в ловушку 2 для отделения от брызг кислоты, затем охлаждается и передается на дальнейшее использование. Когда кислотность раствора в сатураторе снижается до 6-8 % (что соответствует содержанию в нем 140-170 г/л связанного аммиака), из раствора начинают выделяться кристаллы сульфата аммония. Образующаяся пульпа центробежным насосом перекачивается в кристаллоприемник 8. Маточный раствор из верхней части кристаллоприемника переливается в приемный сосуд 6 и возвращается в сатуратор. Кристаллы сульфата аммония непрерывно поступают из кристаллоприемника в центрифугу 7, где отделяются от маточного раствора. Отфугованные кристаллы сульфата аммония, имеющие влажность около 2 %, передают на склад или направляют па сушку.
Часть раствора непрерывно циркулирует между сатуратором и баком 3. Благодаря циркуляции и непрерывному перекачиванию пульпы из сатуратора в кристаллоприемник с возвратом маточного раствора в сатуратор в нем обеспечивается постоянный.уровень жидкости и ее тщательное перемешивание. Поэтому кристаллы соли все время находятся во взвешенном состоянии, и рост кристаллов происходит равномерно во всей массе раствора.
Содержание свободной серной кислоты в маточном растворе, циркулирующем в сатураторе, должно быть в пределах 6-8 %. При понижении кислотности (до 1-2 %) из раствора выпадают более крупные кристаллы, что может вызвать забивку сатуратора солью; при этом также ухудшается поглощение аммиака из газа. С повышением кислотности раствора увеличивается растворимость в нем сульфата аммония и получаются более мелкие кристаллы. Если в растворе содержится более 11 % кислоты, образуется легкорастворимый в воде бисульфат аммония NH4HSО4.
На получение 1 т сульфата аммония затрачивают: 0,73-0,75 т серной кислоты (100 %-й), 0,26-0,27 т аммиака (содержащегося в 30-35 тыс.м 3 коксового газа), 100-108 МДж электроэнергии, 8 м 3 воды и 2,7-6 т пара.
К недостаткам сатураторного способа, помимо малого размера получаемых кристаллов, сильно пылящих при сушке, относится и большой расход энергии на преодоление гидравлического сопротивления абсорберов. Этих недостатков лишены бессатураторные способы.
В бессатураторных процессах абсорбцию аммиака из коксового газа ведут в полых скрубберах или кислым ненасыщенным раствором сульфата аммония с последующей вакуум-выпаркой на кристалл, или кислым насыщенным раствором с выращиванием образовавшихся мелких кристаллов в кристаллизаторах под атмосферным давлением. Чаще используют первый способ — орошение абсорбера ненасыщенным раствором предотвращает их засоление, а кристаллизация в выпарных аппаратах позволяет регулировать размеры получаемых кристаллов. Схема такого процесса показана на рисунке 4.8.

Рисунок 4.8 — Схема производства сульфата аммония бессатураторным способом с вакуум-выпаркой
Аммиак улавливается в полом скруббере 2, снабженном форсунками. Скруббер разделен на две ступени. Нижняя его часть орошается раствором, содержащим 3-4 % свободной Н2SО4, верхняя — раствором, содержащим 10 — 12 % Н2SО4. Коксовый газ из скруббера проходит через ловушку брызг 1 и направляется на дальнейшую переработку. Серная кислота и вода (необходимая для разбавления и компенсации испарения) поступают в сборник 4 раствора, циркулирующего через верхнюю часть скруббера с помощью насоса 5. Часть этого раствора через смолоотделитель 3 подается в сборник 11 маточного раствора, циркулирующего через нижнюю часть скруббера с помощью насоса 12. Сюда же поступает маточный раствор с центрифуги 8.
Из нижней зоны скруббера часть раствора, в котором содержится около 1 % свободной серной кислоты и 40 % сульфата аммония, отбирается через смолоотделитель 3 в сборник 10 и насосом 9 подается в вакуум-выпарной аппарат 6. Образовавшиеся здесь кристаллы опускаются в нижнюю коническую часть аппарата, выполняющую роль кристаллорастителя, где мелкие кристаллы поддерживаются во взвешенном состоянии в восходящем потоке свежего раствора. Это обеспечивает их рост при небольшом пересыщении раствора, и более 60 % кристаллов сульфата аммония получаются с размерами, превышающими 0,5 мм. Такие же результаты достигаются при использовании выпарных аппаратов, снабженных выносными кристаллорастителями. Суспензия из выпарного аппарата, содержащая 50-60 % кристаллов, подается для фильтрования на центрифугу 8, где кристаллы промываются горячим конденсатом при температуре 70-80 °С для удаления остатков кислоты. Затем продукт направляется на сушку.
Производство нитрата калия
Нитрат калия (калиевая селитра) КNО3 — кристаллический бесцветный порошок. Технический продукт может иметь сероватый оттенок. Нитрат калия используют в производстве дымного (черного) пороха, в пиротехнике, в пищевой и стекольной промышленности. Он является ценным безбалластным удобрением, содержащим два питательных элемента — азот и калий (теоретическое содержание — 13,85 % азота и 46,5 % оксида калия). Другие преимущества этого удобрения — малая гигроскопичность и физиологическая щелочность. В основном его применяют в промышленности, так как стоимость азота и калия в нитрате калия больше, чем в других комплексных удобрениях.
Получение нитрата калия нейтрализацией азотной кислоты или при абсорбции оксидов азота гидроксидом или карбонатом калия применяют редко из-за дефицитности и высокой стоимости щелочных реагентов. Наибольшее промышленное распространение имеют конверсионные способы получения нитрата калия из хлорида калия и нитратов натрия, аммония, кальция. Например, при использовании хлорида калия и натрата кальция процесс возможно осуществить методом катионного обмена, попеременно обрабатывая катионит растворами нитрата кальция (с получением раствора нитрата калия) и хлорида калия (регенерация катионита). Далее раствор нитрата калия упаривают, охлаждают, отделяют кристаллы соли на центрифуге и сушат.
Представляет интерес получение нитрата калия из хлорида калия и азотной кислоты или оксидов азота.
4.4.1 Теоретические основы производства нитрата калия конверсионными способами
Наиболее распространен способ, основанный на обменном разложении:
На рисунке 4.9 изображена диаграмма растворимости в водной системе KCl + NaNО3 = NаСl + КNО3 при температурах 5, 25, 50 и 100 о С.

Рисунок 4.9 — Изотермы растворимости в водной системе KCl + NaNО3 = NаСl + КNО3 при температурах 5, 25, 50 и 100 о С
Как видно, при температурах 5-25 °С растворимость солей калия значительно меньше, чем солей натрия (поле кристаллизации нитрата калия занимает большую часть площади квадрата), при температуре 100°С, наоборот, резко увеличивается поле кристаллизации хлорида натрия. Если приготовить раствор эквимолекулярной смеси хлорида калия и нитрата натрия при температуре 100 °С, то фигуративная точка а солевой массы такого раствора, лежащая на пересечении диагоналей квадрата, окажется в поле кристаллизации хлорида натрия. При выпаривании из этого раствора воды при температуре 100 °С, когда будет достигнуто насыщение, начнется кристаллизация хлорида натрия, и состав солевой массы раствора будет изменяться по линии ab.
В точке b раствор станет насыщенным также и хлоридом калия. Если выделившиеся к этому моменту кристаллы хлорида натрия отделить и затем охладить раствор, например до температуры 5 °С то точка b окажется в поле кристаллизации нитрата калия — эта соль и будет выделяться в осадок при охлаждении, причем состав раствора будет изменяться по линии bc.
Так как расстояние между точками а и b невелико, то при выпаривании воды из раствора, содержащего эквимолекулярные количества нитрата натрия и хлорида калия, в осадок выделяется лишь небольшое количество хлорида натрия, и раствор вскоре становится насыщенным также и хлоридом натрия. Это уменьшает и выход кристаллического нитрата калия при охлаждении раствора. Чтобы увеличить количество отделяемого хлорида натрия и повысить выход нитрата калия, как видно из диаграммы, следует вводить в исходный раствор избыток нитрата натрия. Наибольший выход получается, если к концу выделения хлорида натрия раствор насыщен тремя солями — NаСl, КСl и КNО3, т. е. солевая масса его изображается точкой Е2. Тогда после отделения выделившегося хлорида натрия кристаллизация нитрата калия при охлаждении раствора идет по наиболее длинному пути Е2d, что обеспечивает наибольший выход продукта.
Наиболее рациональным и экономичным является осуществление обменного разложения нитрата натрия и хлорида калия в циклическом процессе, когда кристаллизация хлорида натрия идет во время выпаривания из системы воды при постоянном давлении и переменной температуре. Описание и расчет такого оптимального цикла могут быть произведены с помощью комбинации изотермического и изобарического сечений диаграммы. На рисунке 4.10 представлен пример оптимального цикла для случая, когда кристаллизация нитрата калия завершается при температуре 50°С.
Точка а на изотермическом сечении характеризует состав маточного раствора после кристаллизации нитрата калия на отрезке bа в процессе охлаждения. Вначале кристаллизации солевому составу раствора соответствует точка b. Перед кристаллизацией к нему добавляют такое количество воды, чтобы он насытился хлоридом натрия лишь при заданной температуре конца кристаллизации (температура 50 °С). Раствор b получается в результате выпаривания воды и кристаллизации хлорида натрия из кипящего раствора с. Точка с принадлежит к изобарическому сечению диаграммы и лежит на луче выпаривания Всb. Исходный раствор для выпаривания с получается при смешении маточного раствора а с эквимолекулярной смесью хлорида калия и нитрата натрия.

Рисунок 4.10 – Оптимальный цикл конверсии KCl + NaNО3 = NаСl + КNО3 в диаграмме с изотермическим (температура 50°С) и изобарическим (давление 0,1 МПа) сечениями
Таким образом, цикл осуществляется по треугольнику сbа. Чем выше солевой коэффициент цикла (т. е. отношение массы полученного нитрата калия к массе выпаренной воды), тем меньше расход энергии на выпаривание. Чем выше конечная температура кристаллизации нитрата калия, тем меньше затраты на охлаждение раствора. Наиболее экономичными являются циклы с температурными интервалами от точки кипения до конечной температуры кристаллизации, находящейся в пределах температур 50-25 °С. При этом для луча упаривания Вb оптимальные соотношения К + :NO3 — находятся в пределах 0,69-0,96; они обеспечивают сравнительно высокие солевые коэффициенты и небольшие объемы циркулирующих растворов.
4.4.2 Технологическая схема производства нитрата калия
В технике нашли применение только конверсионные способы.
Процесс получения калиевой селитры конверсионным способом (рисунок 4.11) состоит из двух основных стадий:
1) обменное разложение нитрата натрия и хлорида калия;
2) очистка первичной («сырой») калиевой селитры от хлорида натрия и механических примесей.

Рисунок 4.11 – Схема производства калиевой селитры конверсионным методом
Эти две основные стадии включают следующие операции (см. рисунок 4.11):
— растворение хлорида калия в предварительно упаренных растворах нитрата натрия (если в качестве сырья применяется твердый нитрат натрия, то его растворяют до получения раствора заданной концентрации, при этом отпадает процесс упаривания растворов) – проводится в смесителе (мешалка-растворитель) для приготовления раствора исходных солей 1;
— фильтрация смеси растворов хлорида калия и нитрата натрия от нерастворимых примесей (иногда вместо фильтрации ограничиваются отстаиванием смеси) — проводится в фильтр-прессе 2;
— обменное разложение солей — проводится в реакторе 3;
— отделение выпавшего в осадок шлама (хлорид натрия) от раствора нитрата калия — проводится в друк-фильтре или нутч-фильтре 4;
— промывка шлама хлорида натрия и его растворение (полученный раствор хлорида натрия используют в цехах пароснабжения для катионитной очистки воды и других целей) — проводится в сборнике растворов и промывных вод 5 с протеканием реакций разложения примесей:
При этом газообразные продукты разложения примесей вместе с водяным паром, образующимся при проведении процесса конверсии, отводятся в атмосферу;
— кристаллизация нитрата калия из растворов и отделение кристаллов (получение «сырой» соли) – проводится в системе аппаратов «напорный бак первичных растворов нитрата калия 6 – кристаллизатор первичной кристаллизации 7 — центрифуга 8»;
— промывка кристаллов первичной кристаллизации – проводится в центрифуге 8;
— растворение («распарка») кристаллов и отфильтровывание растворов от нерастворимых примесей – проводится в системе аппаратов «растворитель кристаллов «распарник» 9 – фильтр-пресс 4»;
— вторичная кристаллизация нитрата калия из растворов – проводится в кристаллизаторе вторичной кристаллизации 10;
— отделение калиевой селитры от маточного раствора и промывка кристаллов от остатка хлоридов – проводится в центрифуге 12;
— сушка (проводится в сушильном барабане 13) и упаковка готового продукта.
Конверсионный способ производства калиевой селитры имеет крупные недостатки — в качестве сырья применяется дорогостоящий нитрат натрия, для получения которого расходуется дефицитная сода; технологический процесс и аппаратура громоздки и трудоемки, сравнительно велик расход пара.
Производство суперфосфата
Суперфосфат — смесь веществ Ca(H2PO4)2 × H2O и CaSO4. Наиболее распространённое минеральное фосфорное удобрение. Фосфор в суперфосфате присутствует в основном в виде монокальцийфосфата и свободной фосфорной кислоты. Удобрение содержит гипс и др. примеси (фосфаты железа и алюминия, кремнезем, соединения фтора и др.). Простой суперфосфат — серый порошок, почти не слёживаемый, среднерассеваемый; в удобрении 14-19,5 % усвояемого растениями пентаоксида фосфора P2O5.
4.5.1 Теоретические основы процессов
Суперфосфат, который называют также простым суперфосфатом (в отличие от более концентрированного двойного суперфосфата), получают разложением природных фосфатов (апатитового концентрата или фосфоритной муки) серной кислотой. Количество серной кислоты уменьшено по сравнению с необходимым для связывания всего содержащегося в природном фосфате кальция с таким расчетом, чтобы в результате получить смесь монокальцийфосфата и сульфата кальция согласно суммарному уравнению:
В производстве суперфосфата при смешении фосфатного сырья и серной кислоты сначала образуется суспензия, которая по мере 



 протекания химических реакций и кристаллизации постепенно загустевает и твердеет (схватывается) в сплошную массу. Полученный при ее измельчении суперфосфат представляет собой порошок или зерна серого цвета. Он состоит из нескольких твердых фаз и пропитывающей их жидкой фазы. В твердых фазах находятся фосфаты кальция (в основном монокальцийфосфат), магния, железа, алюминия, сульфаи кальция с примесью CaSО4 × 0,5H2O (при переработке апатитового концентрата также сульфат циркония), остатки неразложенных минералов, входящих в состав исходного фосфата, кремнегель SiO2 × nH2O и др. Содержание твердых фаз составляет 65-72 %, в том числе 50-55 % сульфатов кальция и циркония. Жидкая фаза состоит из водного раствора фосфорной кислоты, насыщенного монокальцийфосфатом и содержащего ионы Mg 2+ , Fe 3+ , AI 3+ , F — и др.
протекания химических реакций и кристаллизации постепенно загустевает и твердеет (схватывается) в сплошную массу. Полученный при ее измельчении суперфосфат представляет собой порошок или зерна серого цвета. Он состоит из нескольких твердых фаз и пропитывающей их жидкой фазы. В твердых фазах находятся фосфаты кальция (в основном монокальцийфосфат), магния, железа, алюминия, сульфаи кальция с примесью CaSО4 × 0,5H2O (при переработке апатитового концентрата также сульфат циркония), остатки неразложенных минералов, входящих в состав исходного фосфата, кремнегель SiO2 × nH2O и др. Содержание твердых фаз составляет 65-72 %, в том числе 50-55 % сульфатов кальция и циркония. Жидкая фаза состоит из водного раствора фосфорной кислоты, насыщенного монокальцийфосфатом и содержащего ионы Mg 2+ , Fe 3+ , AI 3+ , F — и др.
Основными операциями в производстве простого суперфосфата являются смешение апатитового концентрата или фосфоритной муки с серной кислотой и отверждение (схватывание) получаемой суспензии в камерах — созревание или вызревание суперфосфата. Окончательное дозревание его происходит при вылеживании и дообработке на складе, который в данном случае в большей мере является химическим цехом, чем хранилищем продукта. Выделяющиеся из смесителей сырья и из суперфосфатных камер фторидные газы улавливаются и перерабатываются на фторсодержащие и другие продукты.
Порошкообразный суперфосфат гигроскопичен и сильно слеживается в результате кристаллизации из жидкой фазы монокальцийфосфата. Меньше слеживается охлажденный и хорошо вызревший суперфосфат, в котором кристаллизация закончилась. Почти не слеживается нейтрализованный и гранулированный суперфосфат.
По мере развития производства концентрированных фосфорных и сложных удобрений удельная доля простого суперфосфата в мировом ассортименте минеральных удобрений уменьшается.
4.5.2 Физико-химические основы получения суперфосфата
Р а з л ожение фосфата. При получении суперфосфата разложение фторапатита протекает в две стадии. Вначале образуется свободная фосфорная кислота:
Эта реакция заканчивается в течение 20-40 мин. в период созревания (твердения) суперфосфатной массы в камере. Сульфат кальция выделяется в осадок в форме полугидрата, но очень быстро, в течение нескольких минут, превращается в ангидрит, в форме которого главным образом и находится в готовом суперфосфате. Это объясняется высокой температурой реакционной массы в суперфосфатной камере (110-120 °С) и большим содержанием пентаоксида фосфора в жидкой фазе (42-46 % в конце первой стадии процесса). При этих условиях стабильной формой сульфата кальция является ангидрит.
После полного израсходования серной кислоты начинается вторая стадия процесса — разложение оставшегося апатита накопившейся фосфорной кислотой по реакции:
Образующийся монокальцийфосфат находится сначала в растворе, при пересыщении которого начинает кристаллизоваться.
При стехиометрическом соотношении компонентов согласно суммарному уравнению:
на первой стадии процесса реагируют 70 %, а на второй — остальные 30 % апатита. На первой стадии образуется структурная сетка из микрокристаллов сульфата кальция, заполненная большим количеством жидкой фазы. Схватывание реакционной массы происходит еще до полного израсходования серной кислоты, в присутствии которой образование монокальцийфосфата невозможно. Поэтому причиной затвердевания массы является только кристаллизация сульфата кальция. Вторая стадия начинается в период камерного созревания и завершается при дозревании продукта на складе в течение длительного времени (6-25 суток, в зависимости от сорта сырья, режима производства и условий дозревания).
Норма серной кислоты. Стехиометрическая норма серной кислоты, расходуемой на разложение фосфата, определяется соотношением 7H2SO4 : 3Р2O5, равным 1,61 ч. серной кислоты на 1 ч. пентаоксида фосфора. При содержании в апатитовом концентрате 39,4 % пентаоксида фосфора стехиометрическая норма серной кислоты составляет 39,4-1,61 = 63,4 ч. на 100 ч. сырья. С целью ускорения разложения и с учетом наличия примесей практическую норму кислоты принимают более высокой — от 68 до 72 ч.
При получении простого суперфосфата из фосфоритов стехиометрическую норму серной кислоты следует рассчитывать с учетом химического и минерального состава сырья и вторичных реакций, протекающих в процессе его разложения по мере уменьшения кислотности реакционной среды. Выделяющиеся кислоты (серная кислота, фтороводород) также разлагают компоненты сырья. Серная кислота расходуется на образование сульфата кальция из содержащегося в сырье кальция, за вычетом той его части, которая идет на получение монокальцийфосфата. Часть фосфатных анионов связывается катионами примесей — Mg 2+ , Fe 3+ , Al 3+ , что вызывает некоторое уменьшение доли кальция, переходящей в монокальцийфосфат, т. е. увеличение количества образующегося сульфата кальция; это требует соответствующего повышения нормы серной кислоты.
Практическую норму серной кислоты для каждого вида сырья обычно устанавливают опытным путем.
Механизм и скорость процесса. Температура и концентрация вводимой в процесс серной кислоты сильно отражаются на структуре и физических свойствах продукта. Скорость разложения фосфата зависит от концентрации серной кислоты, состава и степени пересыщения жидкой фазы суперфосфата продуктами реакции. На рисунке 4.12 показан общий вид зависимости степени разложения фосфата за определенное время (изохрона) от концентрации исходной серной кислоты при периодических условиях смешения.
С увеличением концентрации разбавленных растворов и уменьшением концентрации крепких растворов степень, а следовательно, и скорость разложения фосфата увеличиваются. Однако, начиная с некоторых концентраций кислоты (малых и больших), изменяется состав выделяющегося сульфата кальция и уменьшаются размеры его кристаллов, что вызывает отложение последних на поверхности зерен фосфата и снижение степени разложения. Поэтому полная зависимость степени разложения от концентрации кислоты изображается кривой, которая имеет два максимума и между ними один минимум.

Рисунок 4.12 – Общий вид зависимости степени разложения фосфата от концентрации исходной серной кислоты
Положение максимумов зависит от вида сырья, температуры, времени и др. Скорость и достигаемая степень разложения кислотой низкой концентрации (левый максимум) высоки; но с такой кислотой вводится большое количество воды, и вместо твердого продукта получается несхватывающаяся суспензия.
При разложении апатита в периодических условиях раствором серной кислоты, содержащим выше 63 % серной кислоты, выделяются кристаллы полугидрата сульфата кальция и ангидрита в форме мелких иголочек длиной 5-7 мкм и шириной 1-2 мкм, которые отлагаются на поверхности зерен фосфата, образуя плотный шламовый покров. Это затормаживает реакцию, в результате чего суперфосфатная масса плохо схватывается, содержащаяся в ней жидкая фаза остается на поверхности твердых частиц и получается продукт с плохими физическими свойствами, не рассыпчатый, а «мажущий». При содержании серной кислоты ниже 63 % выделяются более крупные кристаллы сульфата кальция (10-15 мкм). Они образуют пористую рыхлую корку на зернах фосфата, в меньшей степени затрудняющую диффузию к ним кислоты; поэтому реакция идет быстро, и получается сухой рассыпчатый продукт.
Повышение концентрации исходной серной кислоты целесообразно, так как при этом влажность продукта уменьшается и возрастает содержание в нем пентпоксида фосфора. Однако, чрезмерное повышение концентрации серной кислоты, как уже упомянуто выше, вызывает образование на зернах фосфата плотных непроницаемых корок сульфата кальция и снижение скорости разложения сырья. Таким образом, существует область оптимальных концентраций кислоты, границы которой зависят от температуры и условий смешения. Оптимальные концентрации, температура и норма серной кислоты устанавливаются для каждого сорта фосфатного сырья опытным путем.
В промышленном процессе при непрерывном смешении реагентов, которые вводятся в уже частично прореагировавшую реакционную смесь — суспензию (пульпу), используют 68-69 %-ю серную кислоту с температурой 50-60 °С. В этом случае в жидкой фазе устанавливается определенное соотношение серной и фосфорной кислот, условия кристаллизации сульфата кальция улучшаются, и схватившаяся (затвердевшая) масса получается рых- лой.
Продолжительность пребывания суспензии в смесителе должна быть небольшой во избежание загустевания и потери текучести, но достаточной для достижения определенной степени разложения сырья. Чем выше начальная концентрация серной кислоты, тем большая степень разложения апатита должна поддерживаться в суспензии, вытекающей из смесителя, и тем меньше должно быть отношение H2SO4:H3PO4 в жидкой фазе, чтобы на зернах фосфата не образовывались непроницаемые корки сульфата кальция. При начальном содержании серной кислоты 68 % оптимальное время пребывания суспензии в смесителе составляет 5-7 мин, ее температура на выходе из смесителя — 110-115 °С.
Первая стадия разложения фосфата — его взаимодействие с серной кислотой — протекает быстро, тем более, что вначале расходуются мелкие частицы фосфата, а активность раствора 
 (концентрация ионов водорода) велика. Во второй стадии фосфат разлагается фосфорной кислотой, и по мере увеличения степени ее нейтрализации активность раствора и скорость процесса уменьшаются. Когда жидкая фаза становится насыщенной монокальций-фосфатом, что совпадает с окончанием созревания суперфосфатной массы в камере, скорость разложения еще больше замедляется. Это обусловлено, помимо уменьшения активности жидкой фазы, тем, что во второй фазе процесса разлагаются наиболее крупные зерна фосфата, на поверхности которых могут быть шламовые покровы из кристаллов сульфата кальция.
(концентрация ионов водорода) велика. Во второй стадии фосфат разлагается фосфорной кислотой, и по мере увеличения степени ее нейтрализации активность раствора и скорость процесса уменьшаются. Когда жидкая фаза становится насыщенной монокальций-фосфатом, что совпадает с окончанием созревания суперфосфатной массы в камере, скорость разложения еще больше замедляется. Это обусловлено, помимо уменьшения активности жидкой фазы, тем, что во второй фазе процесса разлагаются наиболее крупные зерна фосфата, на поверхности которых могут быть шламовые покровы из кристаллов сульфата кальция.
Уменьшение поверхности контакта фаз и замедление обменной диффузии между ионами водорода и кальция, лежащей в основе кислотного разложения фосфатов, являются главными причинами торможения процесса. Последний полностью прекращается после того, как жидкая фаза суперфосфатной массы становится насыщенной двумя солями — моно- и дикальцийфосфатом. Образование геля, основным компонентом которого является дикальцийфосфат, приводит к появлению на зернах фосфата пленок, полностью подавляющих их взаимодействие с фосфорной кислотой. По указанным причинам дальнейшее вызревание суперфосфата идет очень медленно, уже во время хранения на складе. Возможность его ускорения и степень завершенности процесса, т. е. достигаемая степень разложения сырья, связаны с регулированием фазового состава фосфатного комплекса суперфосфата, т. е. состава смеси из жидкой и твердой фаз, представленных фосфорной кислотой и фосфатами кальция.
Фазовый состав и дозревание суперфосфата. По окончании первой стадии процесса степень нейтрализации равна нулю — жидкая фаза представлена раствором фосфорной кислоты. Во второй стадии, когда фосфорная кислота реагирует с оставшимся фосфатом, жидкая фаза, состоящая из раствора фосфорной кислоты и монокальцийфосфата, может находиться в равновесии с твердым моно- или дикальцийфосфатом. С повышением степени разложения фосфата степень нейтрализации растет, а с увеличением исходной нормы серной кислоты она уменьшается (так как большая часть кальция связывается в сульфат).
Вызревание суперфосфата, полученного из апатитового концентрата, ускоряется при понижении температуры до 40-50 °С. Это объясняется тем, что при охлаждении внешнего слоя жидкости, обволакивающей частицы неразложившегося фосфата, кристаллизуется монокальцийфосфат, насыщающий раствор, и увеличиваются как температурный, так и концентрационный градиенты — реакция ускоряется. Кроме того, в результате кристаллизации Са(Н2РO4)2 × Н2O степень нейтрализации остающейся жидкой фазы уменьшается, а следовательно, активность ее увеличивается, одновременно происходит передвижение жидкой фазы (микроперемешивание). При охлаждении устраняется также возможность возникновения пассивирующих пленок дикальцийфосфата, а если они уже образовались, пока суперфосфатная масса была в камере, то создаются условия для их растворения. Все это способствует ускорению процесса дозревания суперфосфата. Охлаждают суперфосфат, распыляя его в воздухе при подаче из камеры в склад и периодически перебрасывая (перелопачивая) на складе с помощью грейферов или экскаваторов. Происходящее при этом испарение влаги также способствует охлаждению и доразложению.
При оптимальных условиях производства суперфосфата из апатитового концентрата (норма 68 %-й серной кислоты – 69-70 частей., температура 55 °С, продолжительность смешения реагентов 6 мин., время вызревания в камере 1 ч) среднесуточный прирост усвояемого пентаоксида фосфора в первые 5 суток хранения суперфосфата при температуре 40-50 °С составляет 0,3 %, в следующие 5 суток — около 0,1 % и в следующие 10 суток — 0,045 %. Общий прирост усвояемого пентаоксида фосфора за 20 суток достигает 2 %.
П о к а з а т е л и п р о и з в о д с т в а. О полноте разложения фосфатного сырья при производстве суперфосфата судят по коэффициенту разложения — отношению количества усвояемого пентаоксида фосфора к общему его количеству в суперфосфате. Коэффициент разложения тем больше, чем больше норма серной кислоты. При норме 68-72 частей коэффициент разложения в камерном суперфосфате из апатитового концентрата равен 83-88 % и суперфосфат содержит значительное количество свободной фосфорной кислоты (11-12 %). После складского дозревания коэффициент разложения достигает 93-95 %, а содержание свободной фосфорной кислоты снижается до
Отношение количества полученного суперфосфата к количеству затраченного на его производство фосфата называется выходом суперфосфата. Так как содержащийся в фосфате пентаоксид фосфора целиком переходит в суперфосфат, то выход может быть выражен отношением процентного содержания пентаоксида фосфора в фосфате к общему процентного содержания пентаоксида фосфора в продукте. Выход свежеприготовленного суперфосфата из апатитового концентрата составляет 1,94-2,01, из фосфоритов — 1,5-1,9. При хранении на складе выход несколько понижается вследствие испарения части воды.
Г и г р о с к о п и ч н о с т ь п р о д у к т а. Суперфосфат с высокой свободной кислотностью (более 5 % пентаоксида фосфора) при температуре 20 °С в тех случаях, когда относительная влажность воздуха составляет 70-100 %, поглощает атмосферную влагу. Это объясняется небольшим (1,33-1,81 кПа) давлением водяного пара над насыщенными растворами монокальцийфосфата в фосфорной кислоте (жидкая фаза суперфосфата) — оно ниже парциального давления паров воды при той же температуре и соответствующих значениях влажности атмосферного воздуха (1,63-2,33 кПа). Поглощенная влага растворяет некоторое количество монокальцийфосфата, вызывая его разложение на дикальцийфосфат и фосфорную кислоту. Выделение же дополнительного количества свободной фосфорной кислоты еще больше увеличивает гигроскопичность суперфосфата.
Такой суперфосфат обладает плохими физическими свойствами — слеживается, замазывает высевные устройства, зависает в бункерах механических сеялок; кроме того, он вызывает коррозию механизмов и тары, уменьшает всхожесть семян при совместном внесении. Поэтому свободную кислотность вызревшего суперфосфата нейтрализуют, обрабатывая его добавками, легко разлагаемыми фосфорной кислотой. Обычно нейтрализацию сов 

 мещают с гранулированием. В качестве добавок применяют фосфоритную муку из наиболее легко разлагаемых фосфоритов, костяную муку, известь, мел, известняк, обесфторенные фосфаты и др. Добавки, содержащие фосфор, обогащают продукт. Так, при внесении фосфорита снижение содержания свободного пентаоксида фосфора на 1 % эквивалентно приросту усвояемого пентаоксида фосфора в среднем на 0,42 %. При нейтрализации фосфорной кислоты суперфосфата соединениями кальция образуется дополнительное количество монокальцийфосфата. Содержание твердых компонентов в суперфосфате при этом увеличивается, и его физические свойства улучшаются. Однако чрезмерное снижение кислотности суперфосфата может привести к образованию малорастворимого в воде дикальцийфосфата, а избыток извести — к ретроградации с образованием трудноусвояемого трикальцийфосфата. К местной ретроградации может привести и плохое перемешивание реагентов. Поэтому требуется тщательно смешивать суперфосфат с добавками.
мещают с гранулированием. В качестве добавок применяют фосфоритную муку из наиболее легко разлагаемых фосфоритов, костяную муку, известь, мел, известняк, обесфторенные фосфаты и др. Добавки, содержащие фосфор, обогащают продукт. Так, при внесении фосфорита снижение содержания свободного пентаоксида фосфора на 1 % эквивалентно приросту усвояемого пентаоксида фосфора в среднем на 0,42 %. При нейтрализации фосфорной кислоты суперфосфата соединениями кальция образуется дополнительное количество монокальцийфосфата. Содержание твердых компонентов в суперфосфате при этом увеличивается, и его физические свойства улучшаются. Однако чрезмерное снижение кислотности суперфосфата может привести к образованию малорастворимого в воде дикальцийфосфата, а избыток извести — к ретроградации с образованием трудноусвояемого трикальцийфосфата. К местной ретроградации может привести и плохое перемешивание реагентов. Поэтому требуется тщательно смешивать суперфосфат с добавками.
Одним из способов улучшения качества суперфосфата является его аммонизация — нейтрализация свободной кислотности аммиаком. Аммонизированный суперфосфат представляет собой сухой негигроскопичный, неслеживающийся порошок. Содержащийся в нем азот — полезный питательный элемент.
4.5.3 Технологическая схема производства суперфосфата
Суперфосфатный цех включает: склад фосфатного сырья и хранилища серной кислоты; операционное отделение, в котором фосфат разлагают серной кислотой (там же производится поглощение выделяющихся фторидных газов); склад суперфосфата, где продукт подвергается дообработке и дозревает во время вылеживания. Схема производства суперфосфата непрерывным способом изображена на рисунке 4.13.
Поступающий на завод апатитовый концентрат (или фосфоритную муку) выгружают из железнодорожных вагонов и с помощью различных транспортных механизмов или пневматическим способом подают в склад сырья шатрового или силосного типа, а из него – через транспортер для апатитового концентрата 1 в расходный бункер 2 и далее с помощью шнекового питателя 3, ковшевого элеватора 4, шнека 5 (в системе имеется обратный шнек для избыточного апатитового концентрата) в бункер дозатора 7. Во избежание зависания материала в силосах осуществляют аэрацию — через пористые плитки, уложенные в днище силосов, подают сжатый воздух. Система «бункер контрольных весов 12 – контрольные весы для проверки весового дозатора 11» предназначена для проверки работы весового дозатора 8.

Рисунок 4.13 – Схема производства суперфосфата
Серная кислота через резервуар серной кислоты 13 центробежным кислотным насосом 14 непрерывно подается в напорный бак для кислоты 15, где в кислотном смесителе 16 разбавляется водой, подаваемой из напорного бака для воды 17; разбавление контролируется концентратомером 19 по плотности кислоты. Затем через щелевой расходомер кислоты 20 она поступает на смешение с апатитовым концентратом. В случае выделения газов при разбавлении предусмотрен газоотделитель для оксидов азота 18.
Для смешения апатита с кислотой применяют вертикальные трех- или четырехкамерные смесители непрерывного действия 10. Объем суспензии (пульпы) регулируют так, чтобы обеспечить продолжительность ее перемешивания в течение 5-7 минут. Из смесителя суспензия перетекает
в суперфосфатную камеру 21. Она представляет собой вертикальный железобетонный цилиндрический корпус. Камера вращается вокруг оси, в течение 1-2 ч камера делает один оборот.
По мере вращения камеры суперфосфатная масса схватывается и подходит к фрезеру 22 камеры готовой для выгрузки. За один оборот фрезер срезает слой суперфосфата толщиной 5-25 мм. Срезанный суперфосфат попадает на транспортер камерного суперфосфата 23, передающий продукт на склад.
При подаче на склад суперфосфат несколько охлаждают, разбрасывая его. Для этой цели применяют небольшой быстро вращающийся горизонтальный барабан-разбрасыватель 24. Падая на наружную поверхность барабана, суперфосфат отбрасывается лопастями и разбивается на мелкие частицы, которые, оседая в кучу, охлаждаются окружающим воздухом с температуры 70-90 до температуры 30-60°С. Суперфосфат вылеживается в нескольких кучах. Обычно склад оборудован грейферными кранами или другими транспортными механизмами, с помощью которых в течение срока хранения продукт периодически перелопачивают, т. е. пересыпают кучи с места на место. Это способствует его охлаждению и более быстрому дозреванию.
Несмотря на это, срок вылеживания суперфосфата на складе составляет обычно 2-3 недели. При значительной производительности суперфосфатных заводов требуются очень большие склады, намного превышающие по своим габаритам производственные цехи (операционные отделения). В связи с этим задачей суперфосфатной промышленности является переход на способы производства без складского дозревания. Это имеет и важное экологическое значение, так как суперфосфатные склады — источники загрязнения окружающей среды фторидными газами, улавливание которых при малой их концентрации из больших объемов воздуха — задача мало реальная. Получение простого суперфосфата без складского дозревания возможно камернопоточным и поточным способами.
Расходные коэффициенты на 1 т порошкообразного суперфосфата составляют 0,53-0,55 т апатитового концентрата и 0,37-0,38 т серной кислоты (100 %-й).
1. Технология связанного азота/ В.И. Атрощенко, А.М. Алексеев, А.П. Засорин и др..; Под ред. В.И. Атрощенко. — К.: Вища школа, Головное изд-во, 1985. — 327 с: ил.
2. Атрощенко В.И., Каргин С.И. Технология азотной кислоты. — Изд. 3-е, перераб. и доп. — М: Химия, 1970. — 496 с: ил.
3. Амелин А.Г. Технология серной кислоты. Учебное пособие для вузов. -2-е изд., перераб. — М.:Химия, 1983.- 360 с: ил.
4. Ганз С.Н. Синтез аммиака. — К.: Вища школа, Головное изд-во, 1983. – 280 с: ил.
5. Крашенинников С.А. Технология соды: Учеб. пособие для вузов. — 2-е изд., перераб. и доп. — М.: Химия, 1988. -304 с.: ил,
6. Зайцев И.Д., Ткач Г.А., Стоев Н.Д. Производство соды. — М.: Химия, 1986. — 312 с: ил.
7. Семенова Т.А., Лейтес И.Л. и др. Очистка технологических газов. — 2-е изд., перераб. и доп.- М.: Химия, 1977. — 488 с: ил.
8. Позин М.Е. Технология минеральных удобрений: Учебник для вузов. — 6-е изд., перераб. — Л.: Химия, 1989.- 352 с.:ил.
9. Соколов Р.С. Химическая технология: Учеб. пособие для студ. высш. учеб. заведений: В 2 т. — М.: Гуманит. изд. центр ВЛАДОС, 2000. -Т.1: Химическое производство в антропогенной деятельности. Основные вопросы химической технологии. Производство неорганических веществ. — 368 с: ил.
10. Химическая технология неорганических веществ: Учебное пособие для вузов/ Под ред. Ахметова Т.Г. — М.: Химия, 1998. — 488 с: ил.
Источник