Научная электронная библиотека

Юров И. Ю., Воинова В. Ю., Ворсанова С. Г., Юров Ю. Б.,
4.3. Наследование, сцепленное с полом
Х-сцепленное наследование принято делить на Х-сцепленное рецессивное и Х-сцепленное доминантное.
Х-сцепленное рецессивное наследование
Поскольку мужчины имеют только одну хромосому Х, они являются гемизиготными по Х-сцепленным генам. Х-сцепленные рецессивные болезни проявляются у мальчиков, которые имеют только один мутантный аллель, а передаются здоровыми гетерозиготными женщинами-носительницами их сыновьям. Пораженные мужчины, в свою очередь, передают мутантный ген своим дочерям – облигатным носительницам, но не сыновьям. Этот тип передачи в родословной иногда называют «диагональным» (рис. 12).
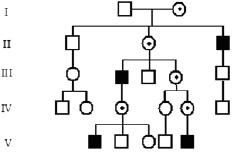
Рис. 12. Родословная при Х-сцепленном рецессивном типе наследовании
Генетический риск. Если облигатная носительница Х-сцепленной рецессивной мутации вступает в брак со здоровым мужчиной, то каждый их сын будет иметь 50 %-й риск заболевания, а каждая дочь – 50 %-й риск быть носительницей. Поскольку мужчина передает хромосому Х только своим дочерям, а хромосому Y – сыновьям, то все дочери пораженных мужчин от браков со здоровыми женщинами являются облигатными носительницами, а все их сыновья здоровы. Таким образом, мужчина не может передать Х-сцепленное заболевание своему сыну за очень редким исключением при унипарентальной гетеродисомии.
В качестве примера Х-сцепленного рецессивного заболевания можно привести мышечную дистрофию Дюшена. Это самая частая мышечная дистрофия, первыми признаками которой являются переваливающаяся походка, трудности при подъеме по лестнице без болевых ощущений и тенденция к падениям ребёнка при ходьбе. Мышечная слабость
прогрессирует и пораженные мальчики умирают в конце второго – начале третьего десятилетия жизни. Таким образом, пораженные мужчины не имеют детей и не передают соответствующие мутации потомкам (рис. 13).
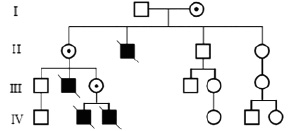
Рис. 13. Родословная семьи с мышечной дистрофией Дюшена
Вариабельная экспрессивность у женщин-гетерозигот. При многих Х-сцепленных болезнях женщины-гетерозиготы имеют мозаичный фенотип. Например, при Х-сцепленном альбинизме радужная оболочка и глазное дно больных мужчин не имеют пигмента, а у гетерозиготных женщин выявляется мозаичная (пятнистая) пигментация. Это объясняется феноменом Х-инактивации.
Х-сцепленное доминантное наследование
Х-сцепленные доминантные болезни являются редкими и выявляются у женщин-гетерозигот, а также у мужчин-гемизигот, имеющих мутантный аллель на единственной хромосоме Х. Х-сцепленное доминантное наследование напоминает аутосомно-доминантное. Но есть значимое отличие: пораженные мужчины передают заболевание только своим дочерям, а передача от отца к сыну невозможна (рис. 14). Примером этого типа наследования является витамин Д-резистентный рахит, при котором женщины обычно имеют более легкие формы заболевания, чем мужчины.
При многих Х-сцепленных доминантных болезнях у женщин может наблюдаться мозаицизм проявления болезни. Например, при синдроме Блоха – Сульцбергера (синдром недержания пигмента, тип II) наблюдается мозаичная пигментация кожи. Кроме того, это заболевание, также как синдром Ретта, является примером болезни, летальной для плодов мужского пола.
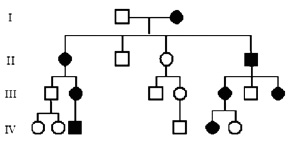
Рис. 14. Родословная при Х-сцепленном доминантном типе наследования
Наследование, сцепленное с хромосомой Y
Наследование, сцепленное с хромосомой Y, предполагает, что болеют только мальчики. Заболевание передается только от отца к сыну. В случае мутаций в генах хромосомы Y, вовлеченных в сперматогенез, возникает бесплодие вследствие азооспермии у мужчин. Технологии искусственного оплодотворения позволяют им иметь детей, но если при этом рождается сын, он также страдает азооспермией.
Влияние пола. Некоторые аутосомные признаки значительно чаще выявляются у одного из полов. Этот феномен получил название влияние пола. Лысина у мужчин является примером аутосомно-доминантного признака, ограниченного полом, что, по-видимому, является результатом влияния мужских половых гормонов. Другой пример – подагра, которая является очень редким состоянием у женщин до менопаузы, но после нее частота этого заболевания возрастает. При гемохроматозе
(аутосомно-рецессивном заболевании) у женщин-гомозигот намного реже возникает перегрузка железом и связанные с ней симптомы, чем у мужчин-гомозигот. Объяснением является физиологическая потеря железа женщинами во время менструаций.
Ограничение полом – это проявление определенных признаков у индивидуумов только одного пола. Пример: вирилизация девочек с аутосомно-рецессивным эндокринным заболеванием – врожденной гиперплазией коры надпочечников.
В табл. 4 кратко представлены основные признаки менделевских типов наследования.
Признаки менделевских типов наследования
Особенности заболевания у лиц разного пола
Особенности передачи
в родословной
Мужчины и женщины болеют в равной пропорции
«Вертикальный» тип передачи – больные во многих поколениях родословной. Передача от лица любого пола лицу любого пола.
Мужчины и женщины болеют в равной пропорции
«Горизонтальный» тип передачи – больные в одном поколении. Родители больного (больных) нередко могут быть родственниками.
Как правило, больны мужчины
«Диагональный» тип передачи: мужчины не могут передавать заболевание своим сыновьям. Возможна передача только внуку от деда через его дочь, которая является непораженой облигатной носительницей.
Болеют мужчины и женщины с преобладанием женщин. Женщины поражены в меньшей степени, чем мужчины. В случае летальных для мальчиков болезней поражены только девочки, наблюдаются спонтанные аборты в семье.
Пораженные мужчины могут передавать заболевание своим дочерям, но не сыновьям. Передача от мужчины к мужчине исключает Х-сцепленный тип наследования.
Сцепленный с хромосомой Y
Болеют только мужчины.
Пораженные мужчины могут передавать заболевание только своим сыновьям.
Множественные аллели и комплексные признаки
Выше рассмотрены признаки, с которыми связаны только два аллеля – нормальный и мутантный. Некоторые гены имеют более двух аллельных форм, т.е. множественные аллели. Некоторые из них могут быть доминантными, другие – рецессивными по отношению к нормальному аллелю. Пример множественных аллелей – наследование групп крови человека.
Развитие генетики сделало возможным исследование комплексных признаков, которые формируются при взаимодействии нескольких генов. На этой основе возникла концепция олигогенного (дигенного и триаллельного) наследования.
При дигенном наследовании наблюдается аддитивный эффект гетерозиготных мутаций в двух различных локусах. Например, одна из форм пигментного ретинита, приводящая к потере зрения, вызвана гетерозиготностью по мутациям двух генов (ROM1 и periferin). Оба эти гена кодируют белки, присутствующие в фоторецепторах сетчатки глаза. Индивидуумы, гетерозиготные по мутации только одного из этих двух генов, не имеют клинических проявлений.
Триаллельное наследование можно рассмотреть на примере синдрома Барде-Бидля – редкого заболевания, характеризующегося ожирением, полидактилией, аномалиями почек, пигментным ретинитом и когнитивными нарушениями. Семь различных генных локусов, мутации в которых ведут к синдрому Барде–Бидля, были идентифицированы. До недавнего времени считалось, что заболевание наследуется аутосомно-рецессивно. Однако, сейчас известно, что есть одна форма синдрома, когда индивидуум, гомозиготный по мутациям одного локуса, является также гетерозиготным по мутации другого локуса. Таким образом, для того, чтобы заболевание проявлялось, необходимо три мутантных аллеля.
Антиципация. При некоторых аутосомно-доминантных болезнях манифестация симптомов более ранняя и течение болезни более тяжелое у потомков по сравнению с их родителями, также страдающими этим заболеванием. Феномен увеличения тяжести болезни из поколения в поколение называют антиципацией. Одним из объяснений антиципации является экспансия нестабильных триплетных повторов. В качестве примеров можно привести такие болезни экспансии триплетных повторов как миотоническая дистрофия, хорея Гентингтона, болезнь Кеннеди.
Источник
Пигментный ретинит
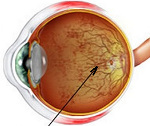
Пигментный ретинит – генетически гетерогенное наследственное заболевание, характеризующееся нарушением функционирования пигментного эпителия сетчатки глаза с развитием разнообразных нарушений. Проявления и выраженность симптомов зависят от формы патологии, наиболее часто наблюдается снижение остроты и сужение поля зрения, развитие скотомы и нарушение темновой адаптации, в дальнейшем может возникать слепота. Диагностика пигментного ретинита производится на основании данных офтальмологических исследований (осмотр глазного дна, электроретинография и электроокулография), молекулярно-генетических анализов. Специфическое лечение этого состояния на сегодняшний день разрабатывается (генная терапия, использование стволовых клеток), в клинической практике применяют поддерживающую терапию.
Общие сведения
Пигментный ретинит (пигментная абиотрофия сетчатки) – наследственное дегенеративное заболевание сетчатки глаза, которое характеризуется развитием выраженных нарушений зрения вплоть до полной слепоты. Это заболевание, как одна из причин потери зрения в различном возрасте, известно с древнейших времен, но термин «пигментный ретинит» был предложен голландским офтальмологом Ф. Дондерсом в 1857 году. По мере развития офтальмологии и генетики удалось выяснить, что это состояние представляет собой целую совокупность заболеваний сетчатки, имеющих различную этиологию, но сходный патогенез. В настоящий момент известно несколько десятков генов и сотни вариантов их мутаций, способных привести к этому заболеванию. Механизм наследования пигментного ретинита также может быть различным – описаны аутосомно-доминантные, аутосомно-рецессивные и сцепленные с Х-хромосомой формы патологии. Среди последних также выделяют рецессивные (болеют только мужчины) и доминантные (поражают лиц обоих полов) разновидности. Усредненное значение встречаемости пигментного ретинита составляет порядка 1:5000, существуют формы заболевания как с большей, так и с меньшей частотой. По данным медицинской статистики, носителями генетических дефектов (включая бессимптомное носительство) являются не менее 100-120 миллионов человек.
Причины и классификация пигментного ретинита
Этиология пигментного ретинита очень разнообразна по причине генетической гетерогенности этого заболевания. В настоящее время выделяют огромное количество форм данного состояния, обусловленных мутациями различных генов. В общих чертах причиной пигментного ретинита являются нарушения метаболизма в фоторецепторах и пигментном эпителии, что приводит к накоплению в сетчатке токсичных побочных веществ. Современная классификация заболевания основывается на механизме наследственной передачи генетического нарушения, по этому критерию определяют четыре основные группы пигментного ретинита.
Пигментный ретинит с аутосомно-доминантным наследованием – является самым распространенным вариантом патологии, по различным данным составляет от 70 до 90% всех случаев заболевания. Причиной этой формы абиотрофии сетчатки могут выступать мутации генов RP1 (8 хромосома), PRPH2 (6 хромосома), RP9 и IMPDH1 (7 хромосома) и целого ряда других. Все эти гены кодируют белки, принимающие участие в метаболизме пигментного эпителия, поэтому нарушения в их структуре ведут к разнообразным расстройствам зрения. Аутосомно-доминантный пигментный ретинит, несмотря на большую встречаемость, характеризуется менее выраженными нарушениями, медленным прогрессированием, что при адекватной поддерживающей терапии в ряде случаев позволяет значительно отсрочить или даже избежать развития слепоты.
Пигментный ретинит с аутосомно-рецессивным механизмом наследования – более редкая форма заболевания. Характеризуется довольно ранним началом, быстрым течением и нередко приводит к полной слепоте в молодом или детском возрасте. Его причина заключается в мутациях генов CRB1 (1 хромосома) и SPATA7 (14 хромосома), также имеются более редкие формы патологии, обусловленные дефектами других генов. Патогенез при аутосомно-рецессивных формах пигментного ретинита изучен недостаточно, предполагается участие белков, кодируемых вышеуказанными генами, в процессах эмбрионального развития органов зрения.
Пигментный ретинит с Х-сцепленным характером наследования – также представляет собой тяжелую форму этого генетического заболевания. Наиболее часто он обусловлен дефектами генов RP2 и RPGR с рецессивным характером наследственной передачи. По этой причине пигментный ретинит такого типа поражает только мальчиков, не имеющих гомологичной Х-хромосомы. Эти гены кодируют белки-ферменты, принимающие активное участие в метаболизме сетчатой оболочки глаза, поэтому их дефект приводит к нарушениям, клинически выражающихся в пигментном ретините.
Пигментный ретинит, обусловленный мутациями митохондриальной ДНК – представляет собой редчайший вариант этого заболевания. Он наследуется только по материнской линии и передается от матери потомству. Врачам-генетикам пока не удалось выявить участки митохондриальной ДНК, которые подвергаются мутации при этой форме патологии.
Существуют также другие типы классификаций этого состояния – по клиническому течению, наличию или отсутствию сопутствующих пороков развития, возрасту наступления патологии (врожденный, ювенильный) и ряду других критериев. В настоящее время единой общепринятой классификации этого состояния нет, однако разделение всех форм заболевания по механизму их наследования считается наиболее удобным и понятным, охватывающим большинство клинических и генетических разновидностей пигментного ретинита.
Симптомы пигментного ретинита
Развитие пигментного ретинита может начинаться в любом возрасте – рецессивные и сцепленные с полом формы заболевания чаще всего возникают еще в раннем детстве, тогда как некоторые аутосомно-доминантные разновидности могут проявлять себя во взрослом и даже пожилом возрасте. Как правило, одним из первых симптомов является снижение темновой адаптации и гемералопия, которая может оставаться единственным проявлением патологии на протяжении нескольких недель (быстропрогрессирующие формы) или лет. При дальнейшем течении пигментного ретинита развивается слепота в ночное время (никталопия) при нормальном уровне дневного зрения. Причиной этих проявлений становится преимущественная дегенерация палочек, отвечающих за световосприятие в условиях пониженной освещенности.
В дальнейшем пигментный ретинит характеризуется сужением поля зрения и выпадения его периферических участков (периферическая скотома). Это также является продолжением разрушения палочек, которые располагаются в основном по краям сетчатки. В тяжелых случаях развивается «туннельное» зрение, его острота значительно падает, больные пигментным ретинитом становятся инвалидами. Дегенеративные изменения затрагивают и сосуды глаза, что ведет к разрушению колбочек, помутнению хрусталика и стекловидного тела, истончению склер. Совокупность этих процессов приводит к полной слепоте больного. Однако далеко не каждая форма пигментного ретинита характеризуется таким исходом – многие аутосомно-доминантные разновидности заболевания длительное время проявляются только гемералопией и незначительным сужением поля зрения.
Диагностика пигментного ретинита
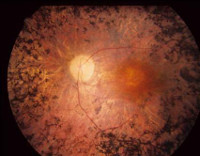
Для выявления пигментного ретинита используют осмотр глазного дна, электроретинографию, электроокулографию и другие офтальмологические исследования, изучение наследственного анамнеза больного, молекулярно-генетические анализы. При жалобах пациента на снижение зрения в вечерние часы необходимо производить полноценный офтальмологический осмотр. На глазном дне могут выявляться отдельные точки (костные пятна), расположенные по периферии сетчатки – они представляют собой отложения жироподобного пигмента. По мере прогрессирования пигментного ретинита их становится все больше, они начинают образовываться ближе к желтому пятну. При выраженной клинической картине заболевания на глазном дне также определяется сужение артериол, атрофия капилляров, а в дальнейшем – восковидная атрофия диска зрительного нерва.
Измерение ширины полей зрения при пигментном ретините обнаруживает их концентрическое сужение различной (в зависимости от стадии заболевания) степени выраженности. Характерным проявлением этой патологии также является снижение чувствительности синего цвета вплоть до тританопии, что определяется при помощи таблиц Рабкина. Картина электроретинографии при пигментном ретините зависит от стадии патологии – начиная от снижения всех волн и заканчивая нерегистрируемой ЭРГ при полной слепоте. Целью проведения электроокулографии является вычисление коэффициента Ардена, который в норме составляет не менее 180%. При пигментном ретините его значение может снижаться до 100% и даже ниже.
Молекулярно-генетические исследования необходимы для окончательного подтверждения диагноза пигментного ретинита, кроме того, эти данные могут быть полезными при определении прогноза заболевания. В настоящее время в лабораториях доступны методы генетической диагностики наиболее распространенных форм патологии, обусловленных мутациями генов RP1, RP2, RPO, CRB1, SPATA7, RPGR и ряда других. Эти исследования охватывают примерно70-80% всех случаев пигментного ретинита, но в отношении многочисленных более редких форм генетические методы диагностики не разработаны. Как правило, диагностическая техника в этом случае сводится к прямому или автоматическому секвенированию последовательности вышеуказанных генов.
Лечение и прогноз пигментного ретинита
В настоящий момент специфические методы лечения пигментного ретинита находятся в стадии разработки и клинических испытаний. Имеются перспективные результаты применения генной терапии, стволовых клеток и других медицинских методик. В клинической практике используют только поддерживающее лечение, направленное на замедление прогрессирования проявлений пигментного ретинита. С этой целью применяют препараты витамина А, средства, улучшающую трофику сетчатки и других структур органов зрения. В некоторых странах разработаны протезы сетчатки, их имплантация положительно влияет на зрительную функцию больных пигментных ретинитом. Однако во многих случаях, особенно при аутосомно-рецессивных и сцепленных с полом формах заболевания, несмотря на все терапевтические мероприятия, развивается необратимая слепота.
Прогноз пигментного ретинита считается в целом неблагоприятным, поскольку заболевание неуклонно прогрессирует, приводя в конечном итоге к полной слепоте. У различных форм этого состояния отличается только скорость нарастания симптомов – она выше у аутосомно-рецессивных разновидностей и значительно ниже при доминантных типах патологии. Поддерживающее лечение способно отсрочить наступление слепоты в среднем на 5-10 лет, но никаких других лечебных мероприятий в клинической практике относительно пигментного ретинита на сегодняшний день не существует. Профилактика возможна в качестве медико-генетического консультирования родителей, входящих в группы риска (больные пигментным ретинитом или его наличие у близких родственников). Также рекомендуется использование солнцезащитных очков, которые, по некоторым данным, замедляют прогрессирование симптомов заболевания.
Источник