Перечислите способы расщепления рацематов
8.5. Методы разделения энантиомеров
Операции разделения рацемических смесей на составляющие их оптически активные компоненты называются расщеплением. Если хотя бы один энантиомер удается выделить в чистом виде, расщепление называют полным, в противном случае говорят, что произошло частичное расщепление, т.е. оптически активное соединение содержит примесь второго энантиомера.
Отношение экспериментально наблюдаемого удельного вращения вещества, полученного путем расщепления, к удельному (абсолютному вращению чистого энантиомера называется оптической чистотой (Р). Тождественными оптической чистоте являются понятия энантиомерной чистоты или энантиомерного избытка (э.и.).
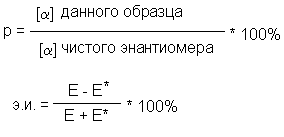
где Е — мольная доля энантиомера, находящегося в избытке,
Е * — мольная доля другого энантиомера.
Любой процесс получения оптически активного вещества из оптически неактивного предшественника, в том числе и расщепление рацемических смесей, называется оптической активацией. Общим принципом всех процессов оптической активации является создание в той или иной форме диастереомерных взаимодействий.
8.5.1. Расщепление через диастереомеры
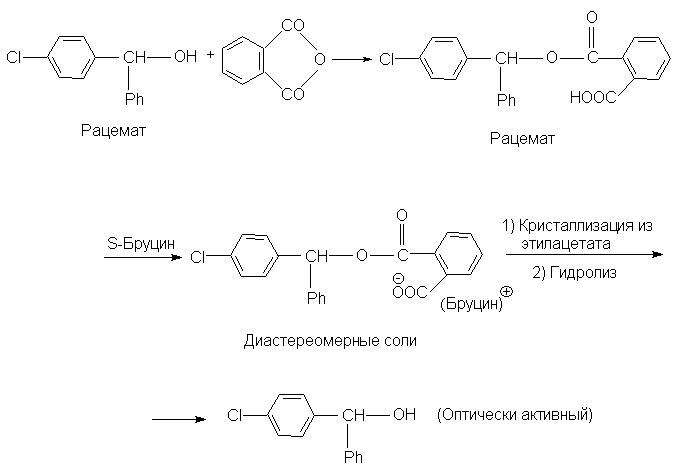
Этот метод до настоящего времени использовался наиболее часто. Если рацемическое соединение содержит карбоксильную группу, то можно получить соль с оптически активным основанием. Если же рацемат содержит аминогруппу, то можно получить соль с оптически активной кислотой. Допустим, что оптически активный реагент (в данном случае основание или кислота) имеет (S)-конфигурацию. Тогда образующиеся соли будут смесью (R)- и (S)-диастереомеров, и в отличие от энантиомеров их свойства будут уже различаться.

В принципе для разделения отличающихся по свойствам диастереомеров можно использовать разные методы, но на практике чаще всего применяют кристаллизацию, т.е. используют различие в растворимости двух диастереомеров. В настоящее время все чаще применяют хроматографические методы. На последней стадии из соли выделяют знантиомер.
Подбор реагентов для разделения данной рацемической смеси производится исключительно эмпирически, т.к. каких-либо теоретических предпосылок для прогнозирования различной растворимости диастереомерных солей не существует.
Для разделения рацемических кислотных соединений применяют природные оптически активные основания, которые называются алкалоидами, например, бруцин, эфедрин, стрихнин, хинин, цинхонин, морфин и др. После проведения разделения их регенерируют и используют снова. Однако эти вещества сильно токсичны и поэтому их стремятся заменить синтетическими оптически активными аминами, например, a -фенилэтиламином. Например, таким путем расщепляется рацемическая 3-метил-2-фенилбутановая кислота.
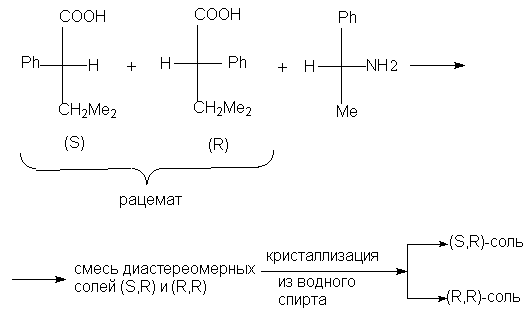
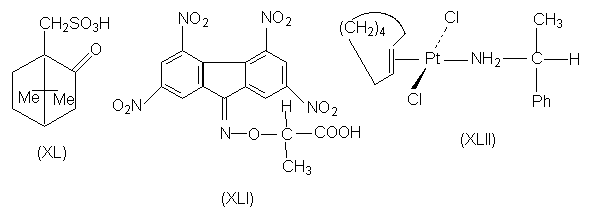
Для разделения рацемических основных соединений применяют оптически активные кислоты: винную, миндальную (a -гидроксифенилуксусную), аспарагиновую (аминоянтарную), глутаминовую (a -аминоглутаровую), камфорсульфоновую (XL)и др.
К сожалению, различия в растворимости энантиомеров редко бывает достаточно велико, для того чтобы осуществить полное разделение в ходе одной операции, обычно приходится проводить многократную кристаллизацию, что делает разделение длительным трудоемким процессом.
Если молекула не содержит кислотной или основной группировки, то ее можно сначала ввести, а затем после разделения на энантиомеры снять, например,
Диастереомеры могут образовываться не только в результате взаимодействий кислот и оснований Бренстеда, как описано выше, но также и в реакциях, в которых взаимодействуют кислоты и основания Льюиса. Так, при расщеплении ароматических соединений, в состав которых не входит ни кислотные, ни основные группировки (например, хиральных нафтиловых эфиров), может быть использована их способность образовывать p -комплексы с нитрофлуореном. Для этой цели используют реагент (XLI), в котором элекктроноакцепторные тетранитрофлуореноноксимная группа придает ей способность к комплексообразованию с электронодонорными ароматическими кольцами, а фрагмент энантиомерной молочной кислоты обеспечивает реагенту в целом оптическую активность. Другим примером является расщепление транс-циклооктена путем образования комплекса с солью двухвалентной платины (кислота Льюиса), вторым лигандом у которой является молекула (R)- a -фенилэтиламина (XLII).
8.5.2. Хроматографическое расщепление
Если рацемичеcкую смесь хроматографировать на колонке, заполненной хиральными веществами, энантиомеры должны проходить с разными скоростями и, следовательно, их можно разделить. Таким путем, например, миндальную кислоту разделяют на колонке, заполненной крахмалом. Можно использовать бумажную, колоночную, газовую и жидкостную хроматографию.
8.5.3. Механическое расщепление
В случае рацемической натрийаммониевой соли винной кислоты энантиомеры при температуре ниже 27 0 (здесь температура очень важна) кристаллизуются раздельно: в одном кристалле собираются (+)-изомеры, а в другом (-)-изомеры. Такие кристаллы отличаются друг от друга зеркальностью формы, и их можно разделить с помощью пинцета и микроскопа. Именно таким путем Л. Пастер в 1848 г. впервые доказал, что рацемическая винная кислота в действительности представляет собой смесь (+)- и (-)-изомеров.
Однако такого рода кристаллизация свойственна лишь немногим веществам. Описано, например, расщепление гептагелицена (смесь спирально сочлененных бензольных колец; аналог гексагелицена — ). Один из энантиомеров этого соединения, имеющий необычно высокое оптическое вращение ([a ]D 20 = +6200 0 ) спонтанно выкристаллизовывается из бензола.
При аналогичном расщеплении 5-метил-3,3-диэтил-2,4-пиперидиндиона (XLIII) было взято 20 кг рацемата и после 400 перекристаллизаций получено всего 3 г оптически чистого правовращающего изомера. Одним из немногих соединений, которые можно разделить пинцетом по методу Пастера является 1,1 , -динафтил (XLIV). При нагревании рацемата при 76-150 0 происходит фазовое изменение с образованием лево- и правовращающих кристаллов.
8.5.4. Ферментативное расщепление
Довольно часто для получения оптически активных веществ из рацематов используют ферменты, которые обладают высокой стереоспецифичностью действия. Наибольшее значение метод приобрел для стереоспецифического гидролиза N-ациламинокислот. Под действием фермента ацилазы на рацемическую N- ацетиламинокислоту L-изомер гидролизуется в 1000 раз быстрее D-изомера, и после окончания ферментативной реакции легко можно разделить L-аминокислоту и D-ацетиламинокислоту.
8.5.5. Установление оптической чистоты
В большинстве случаев при расщеплении рацематов получаются энантиомеры, не имеющие 100%-ной оптической чистоты. Для установления содержания в них второго энантиомера применяют по сути дела те же методы, что и для расщепления, с той лишь разницей, что в данном случае образующиеся диастереомерные комплексы не разделяют, а тем или иным способом определяют их концентрацию. Относительные концентрации диастереомеров можно определить любым способом, например, с помощью ГЖХ или ЯМР-спектроскопии.
8.6. Асимметрический синтез и катализ
Асимметрическим синтезом называют реакции, в ходе которых один из двух энантиомеров хирального продукта образуется в большем количестве, чем второй. В асимметрическом синтезе ключевой является стадия, в которой так называемый прохиральный реагент превращается в хиральный продукт. Прохиральными называются молекулы, способные превратиться в хиральные молекулы путем «одношагового» преобразования структуры. Например, фенилуксусную кислоту в одну стадию можно превратить в a -бромфенилуксусную кислоту, которая хиральна:
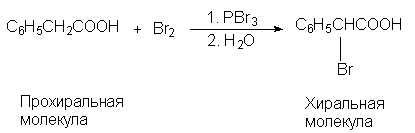
В этой реакции (R)- и (S)-изомерные a -бромфенилуксусные кислоты образуются в строго одинаковых количествах (э.и.=0), и, следовательно, реакцию нельзя назвать асимметрическим синтезом, хотя она и приводит к образованию хирального продукта из ахирального. Чтобы энантиомерный избыток был отличен от нуля, необходимо обязательно соблюсти одно очень важное условие. Это условие состоит в том, что в ходе реакции обязательно должны возникнуть диастереомерые отношения между вновь возникающим хиральным элементом (в рассматриваемом примере — центром хиральности) и вторым хиральным элементом, специально вводимым в реагирующую систему. Например, прохиральная молекула натриевой соли метилэтилмалоновой кислоты при декарбоксилировании дает рацемическую 2-метилмасляную кислоту, но бруциновая соль дает продукт с избытком левовращающего изомера:
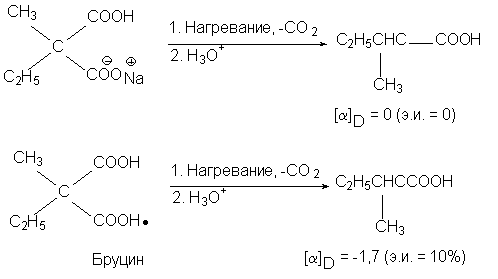
Реакция декарбоксилирования бруциновой соли явилась первым примером асимметрического синтеза (В. Марквальд, 1904). Здесь вторым хиральным элементом является асимметрический центр в молекуле бруцина.
В том же 1904 г А. Маккензи осуществил асимметрические синтезы, в которых исходным реагентом служил (-)-ментиловый эфир фенилглиоксиновой кислоты (XLV).
В этом примере вторым хиральным элементом является хиральный центр в ментиловой группе.
Глубокий смысл необходимости присутствия второго хирального элемента состоит в том, что Левое будет более предпочтительно, чем Правое (или Правое более предпочтительно, чем Левое) лишь в том случае, когда имеется второй элемент, который тоже может быть левым или правым, и поэтому «распознает» энергетическую разницу между подходом реагента (СН3MgI во втором примере) слева или справа, и способствует определенной наиболее выгодной ориентации.
В рассмотренных выше примерах вспомогательный хиральный элемент содержался в самом субстрате. Однако стереохимический результат реакции зависит не от симметрии одного лишь реагента, а от полной симметрии реагирующей системы. Поэтому при проведении асимметрического синтеза используют (1) хиральные субстраты, содержащие прохиральные группы, (2) хиральные реагенты (например, хиральные гидриды при гидрировании кратных связей), (3) хиральные катализаторы и (4) хиральные растворители. Стереоселективность (энантиомерный избыток) асимметрического синтеза колеблется в широких пределах, достигая 98% при использовании некоторых хиральных катализаторов (см. ниже, а также гл. 27), а в ферментативных реакциях даже 100%. Реакции с селективностью 100% называются стереоспецифическими.
Современные представления о механизме асимметрического синтеза целиком основаны на конформационном анализе. Но прежде чем перейти к механизму, необходимо еще раз и более детально рассмотреть вопросы внутримолекулярной симметрии, поскольку многие молекулы содержат атомы или группы атомов, которые лишь кажутся эквивалентными, но при строгой проверке на самом деле оказываются разными.
Сервер создается при поддержке Российского фонда фундаментальных исследований
Не разрешается копирование материалов и размещение на других Web-сайтах
Вебдизайн: Copyright (C) И. Миняйлова и В. Миняйлов
Copyright (C) Химический факультет МГУ
Написать письмо редактору
Источник
III. Методы получения стереоизомеров
Получение чистых стереоизомерных форм — важная часть многих стереохимических исследований. Между тем, в обычных условиях образуются, как правило, смеси стереоизомеров — цис-/транс-форм, диастереомеров или оптических антиподов. Для получения чистых стереоизомерных форм эти смеси необходимо разделять.
Так как цис-/транс-изомеры, а также диастереомеры различаются по физическим свойствам, разделение их смесей обычно не вызывает затруднений. Для этого используют различия в растворимости(разделение перекристаллизацией), различия в температурах кипения (разделение перегонкой), различия в адсорбционной способности (разделение с помощью различных хроматографических методов).
Хорошо известно, например, что геометрические изомеры 1,2-этилендикарбоновой кислоты — фумаровая (транс-форма) и малеиновая (цис-форма) кислоты, сильно различаются по растворимости в воде: растворимость малеиновой кислоты в воде почти в 100 раз больше растворимости фумаровой. Очевидно, что эти кислоты легко отделить друг от друга перекристаллизацией. Сильно различаются по растворимости и такие диастереомерные вещества, как мезо- и (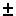 )-формы альфа, бета-дибромглутаровых кислот: кипящим хлороформом можно извлечь из смеси рацемат, а в остатке накопится менее растворимый мезо-изомер.
)-формы альфа, бета-дибромглутаровых кислот: кипящим хлороформом можно извлечь из смеси рацемат, а в остатке накопится менее растворимый мезо-изомер.
В отличие от этого, получение оптически активных веществ — более сложная задача, потому что оптические антиподы не различаются ни по растворимости, ни по температурам кипения, ни по адсорбционному сродству к обычным адсорбентам, т.е. ни перекристаллизацией, ни перегонкой, ни обычными адсорбционными методами пару оптических антиподов разделить нельзя.
Поскольку обычные синтезы приводят к рацематам, возникает вопрос, откуда же берутся оптически активные вещества? Важным источником их является живая природа: оптической активностью обладают белки и составляющие их природные аминокислоты, углеводы, многие природные оксикислоты (винная, яблочная, миндальная), терпеновые углеводороды, терпеновые спирты и кетоны, стероиды, алкалоиды и др. Синтетическое получение оптически активных веществ основано на использовании двух методов:
1. Расщепление рацематов, т.е. отделение друг от друга оптических антиподов, входящих в состав рацемата.
2. Асимметрический синтез — проведение стереоспецифичных реакций, в ходе которых преимущественно образуется ( или преимущественно разрушается) один из антиподов.
Рассмотрим более подробно каждый из этих методов.
III.1. Расщепление рацематов
А. Расщепление рацематов отбором кристаллов и самопроизвольной кристаллизацией
Впервые оптически активное вещество из неактивного получил Пастер в 1848 году путем отбора кристаллов. Он заметил, что из водных растворов натриевоаммониевой соли виноградной кислоты (HOOC-CH(OH)-CH(OH)-COOH) выпадают два типа кристаллов, отличающиеся друг от друга зеркальностью формы. Вручную разделив оба вида кристаллов и приготовив из них водные растворы, Пастер обнаружил, что в отличие от исходной соли эти растворы оптически активны: один вид кристаллов дал левовращающий, а другой правовращающий раствор.
К методу расщепления рацематов отбором кристаллов примыкает расщепление кристаллизацией, т.е. протекающее как бы «самопроизвольно». Чаще всего такого рода «самопроизвольное» выделение одного из антиподов из раствора рацемата удается вызвать, внося в пересыщенный раствор рацемата «затравку» одного из антиподов.
Б. Поведение антиподов в оптически активных растворителях
Путем кристаллизации из оптически активных растворителей удается выделить энантиомерные вещества лишь в тех случаях, когда молекула растворенного вещества и растворителя взаимодействуют минимум в двух точках: для этого они должны иметь не менее двух полярных групп каждая. Так, кристаллизацией из оптически активного диизопропилового эфира винной кислоты удалось расщепить 2,3-дибромбутандиол-1,4 и 1,2-бис-(гамма-пиридил)-этиленгликоль:
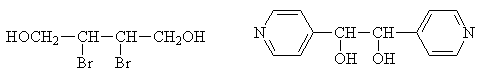
Другой вариант использования оптически активного растворителя — распределение расщепляемого рацемата между ним и оптически неактивной фазой. Так, распределением между водой и эфирами (+)-винной кислоты удалось расщепить 2,3-дибромбутандиол-1,4.
В. Расщепление через диастереомеры.
Способ расщепления через диастереомеры является практически наиболее важным для получения оптически активных веществ: в определенных случаях с ним может конкурировать лишь биохимический метод. Суть способа может быть выражена следующей схемой:
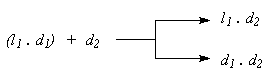
На расщепляемый рацемат ll . d1 действуют оптическим активным реагентом d2: при этом получается новая пара веществ l1 . d2 и d1 . d2, уже не находящиеся друг с другом в отношениях антиподов — эти вещества диастереомерны, а, значит, различаются по физическим свойствам. Разницы в растворимости, давлении пара, коэффициентах адсорбции во многих случаях оказывается достаточно, чтобы разделить диастереомеры кристаллизацией, перегонкой или хроматографически.
На практике расщепление рацематов через диастереомеры включает три последовательные операции: образование пары диастереомеров, их отделение друг от друга, разрушение каждого из диастереомеров, причем выделяются антиподы разделяемого вещества.
Очевидно, что образование диастереомеров возможно только в том случае, если разделяемое вещество имеет химически активную группу, способную взаимодействовать с подходящим асимметрическим реагентом. Природа этой группы, вообще говоря, безразлична. Важно лишь, чтобы при реакции не затрагивались связи асимметрического центра, а образование и расщепление диастереомеров происходило легко. Практически чаще всего используют диастереомерные соли, все остальные реакции имеют несравненно меньшее значение. Для расщепления рацемических кислот необходимы оптически активные основания, для расщепления рацемических оснований — оптически активные кислоты.
Большинство асимметрических реактивов — вещества дорогие, поэтому большое значение имеет возможность регенерации реактивов после окончания расщепления. Пожалуй, единственный реагент, в отношении которого проблема регенерации не стоит из-за его дешевизны — это винная кислота.
На практике наибольшее распространение получили следующие асимметрические реактивы.
1) Для расщепления рацемических карбоновых кислот:
— реагенты основного характера: алкалоиды (хинин, бруцин, стрихнин), альфа -фенилэтиламин, альфа-бензилэтиламин, альфа-(нафтил-1)-этиламин и т.п.
— оптически активные спирты (ментол и т.п.) — в этом случае разделяют диастереомерные сложные эфиры.
2) Для расщепления рацемических аминов — асимметрические реагенты кислотного характера. Как правило, это (+)-винная кислота (наиболее дешевый и доступный из асимметрических реактивов), а также пироглутаминовая кислота I, легко получающаяся при нагревании природной глутаминовой кислоты [ПРИМ.11].
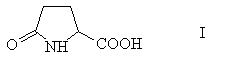
Большая группа асимметрических реагентов кислотного характера получена также из природных терпеновых кетонов и спиртов путем превращения их в сульфокислоты или кислые сульфаты.
Для расщепления рацемических аминокислот можно использовать как солеобразование по карбоксильной группе (после предварительной защиты аминогруппы), так и солеобразование по аминогруппе (после превращения карбоксильной группы, например, в сложноэфирную).
Г .Адсорбционное расщепление.
Оптические антиподы по-разному сорбируются на оптически активном адсорбенте. Так, кварц, способный образовывать хиральные кристаллы, имеет разную адсорбционную способность по отношению к оптическим антиподам; таким образом удалось получить в оптически активной форме ряд неорганических комплексных соединений.
В качестве органических асимметрических адсорбентов используются лактоза, бруцин, крахмал. Можно использовать полимеры, с введенными в состав этого полимера остатками оптически активных аминов или кислот.
Д. Биохимическое получение оптически активных веществ.
Еще в 1857 году Пастером было замечено преимущественное разрушение некоторыми микроорганизмами (например, плесневым грибком Penicillum glaucum) правовращающей формы винной кислоты. Если же действию грибка подвергался рацемат, то не затрагиваемый левовращающий антипод можно было накопить и получить в чистом виде.
Биохимический метод особенно успешно используется для получения оптически активных аминокислот. Так, дрожжи в процессе брожения перерабатывают преимущественно L-формы аминокислот, а D-антиподы накапливаются и могут быть выделены. Под действием фермента ацилазы N-ацетил-L-метионин гидролизуется, например в 1000 раз быстрее чем ацетильное производное D-метионина.
Завершая рассмотрение способов расщепления рацематов, подчеркнем, что во всех случаях оптически активное вещество генерируется только под действием другого оптически активного вещества (реагента, растворителя, адсорбента) [ПРИМ.12] и антиподы проявляют различия в свойствах только при действии других оптически активных веществ (реагентов, растворителей, адсорбентов или плоско поляризованного света).
III.2. Асимметрический синтез
Асимметрический синтез — второй важнейший путь получения оптически активных веществ. Сущность асимметрического синтеза состоит в проведении стереоселективных реакций, в ходе которых антиподы образуются или разрушаются в неравных количествах. Напомним, что расщепление рацематов сводится к разделению антиподов, при асимметрическом синтезе антиподы возникают так, что преобладание одного из них в продуктах реакции приводит к появлению оптической активности [ПРИМ.13].
Различают следующие типы асимметрических синтезов:
1. Частичный асимметрический синтез, осуществляемый при участии вспомогательных оптически активных веществ, созданных живой природой. Разновидностью частичного асимметрического синтеза являются кинетические превращения и кинетические расщепления (см. ниже).
2. Абсолютный асимметрический синтез — процессы получения оптически активных веществ без участия вспомогательных оптически активных веществ или какий-либо иных факторов, зависящих от живой природы.
А. Асимметрические превращения и кинетическое расщепление
Многие оптически активные вещества обладают лишь ограниченной устойчивостью во времени: их оптическая активность постепенно падает и в конце концов исчезает. Такое явление называют рацемизацией [ПРИМ.14].
Чаще всего рацемизация происходит не самопроизвольно, а вызывается какими-либо физико-химическими воздействиями (например, часто вызывают рацемизацию кислотные и щелочные микропримеси). Если процесс изменения конфигурации асимметрического центра совершается под влиянием других элементов хиральности, находящихся в самом веществе или в его окружении (растворитель, катализатор), то момент достижения равновесия не должен непременно совпадать с созданием равномолекулярной смеси обоих антиподов: под воздействием второго хирального центра или хиральной среды одна из форм (в первом случае — один из диастереомеров, во втором — один из антиподов) может оказаться более выгодной, чем другая. В этом случае исходя из рацемата можно получить смеси антиподов с преобладанием одного из них: по существу, речь идет о процессе, обратном рацемизации. Подобные процессы называются асимметрическими превращениями.
Так, при длительном нагревании рацемической миндальной кислоты с бруцином был получена миндальная кислота, обладавшая небольшим оптическим вращением.
При определенных превращениях оптические антиподы реагируют с разными скоростями. Это наблюдается либо при реакциях, протекающих в присутствии оптически активных катализаторов, либо при реакциях с оптически активными веществами. Если в подобную реакцию ввести рацемат, и превращение прервать до его полного завершения, то один из антиподов, реагируя быстрее, будет преобладать в продукте реакции, второй — в непрореагировавшем остатке. Таким образом можно добиться кинетического расщепления. Кинетическому расщеплению подвергается рацемическая миндальная кислота в реакции с ментолом: соотношение скоростей реакций (-)- и (+)-форм равно 0.897.
Б. Возникновение асимметрического центра из карбонильной группы
Частичные асимметрические синтезы с образованием нового центра асимметрии часто осуществляются путем превращения карбонильной группы во вторично-спиртовую или третично-спиртовую по схемам:
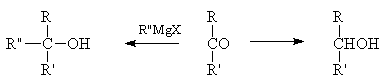
Приведем некоторые примеры синтезов на основе этой схемы.
1. Одним из излюбленных объектов для проведения асимметрического синтеза является бензоилмуравьиная (фенилглиоксиловая) кислота С6H5COCOOH. Для нее и для других кетокислот в принципе возможны два варианта проведения асимметрических синтезов: вспомогательное оптически активное вещество может быть введено либо в молекулу кислоты (чаще всего до сих пор использовались эфиры с ментолом), либо асимметризующее действие оказывает используемый оптически активный реагент.
Первый вариант можно иллюстрировать получением оптически активной миндальной кислоты из эфира, образующегося при восстановлении ментилового эфира бензоилмуравьиной кислоты:
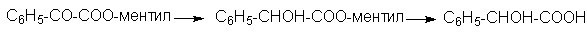
При действии на то же исходное вещество магнийорганических соединений идет асимметрический синтез с образованием третично-спиртовой группы:
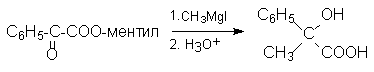
2. При реакции оптически активного ментилмагнийбромида на фенилглиоксиловую кислоту удается получить миндальную кислоту оптической чистоты 35%. Эта реакция представляет собой пример участия в асимметрическом синтезе оптически активного реагента:
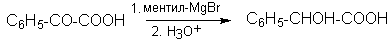
В. Присоединение по двойной связи С=С
Реакции присоединения по двойной углерод углеродной связи — важная разновидность частичных асимметрических синтезов. Примеры:
1. Каталитическое гидрирование непредельных кислот в виде эфиров с оптически активными спиртами.
2. Реакции гидроборирования с помощью реагента, получаемого из (-)-пинена:
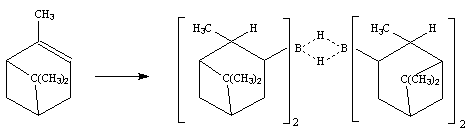
Это один из наиболее стереоспецифичных реагентов в реакциях присоединения. При его действии на 1,2-цис-дизамещенные этилены получаются продукты гидроборирования, которые превращаются в оптически активные спирты с оптической чистотой, близкой к 100%.
3. Присоединение брома по двойной углерод-углеродной связи в присутствии алкалоидов приводит к оптически активным дибромидам.
Г. Синтезы на асимметрических катализаторах
Перспективный способ получения оптически активных веществ — проведение реакций (главным образом, гидрирования) в присутствии катализаторов, обладающих асимметрическим действием. Так, при использовании в качестве асимметрического катализатора палладия, нанесенного на фиброин шелка удается стереоселективно прогидрировать C=N cвязи оксимов. Можно также придать скелетному никелевому катализатору асимметризующую способность, обработав его перед проведением гидрирования оптически активном веществом.
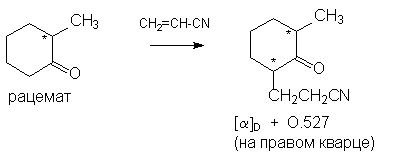
Рассмотренные выше примеры каталитического получения оптически активных веществ относятся к области частичного асимметрического синтеза, поскольку требуют для своего осуществления вспомогательных природных асимметризующих реагентов. Существует и возможность абсолютного асимметрического синтеза при проведении катализа на дисимметричных кристаллах, например, оптически активных кристаллах кварца. Так, при проведении реакции асимметрического цианэтилирования под влиянием нанесенного на кварц щелочного катализатора в мягких условиях (комнатная температура) образуются оптически активные продукты, например:
Д. Абсолютный асимметрический фотохимический синтез
В области оптически активных полос поглощения наблюдается круговой (циркулярный) дихроизм — неравенство коэффициентов поглощения правого и левого циркулярно поляризованного света оптическими антиподами. Один из антиподов сильнее поглощает одну компоненту циркулярно-поляризованного света, другой антипод — другую. Таким образом, если рацемат освещать однородным (правым или левым) циркулярно поляризованным светом, то один из антиподов будет поглощать больше световой энергии, чем другой, а поскольку именно поглощенный свет, согласно основному закону фотохимии, может вызывать химические изменения, описанное явление создает основу для проведения асимметрических синтезов под влиянием циркулярно-поляризованного света.
Например, присоединение галогена к триарилметильному радикалу под действием циркулярно-поляризованного света дает продукт реакции, имеющий вращение до 0.2 о :
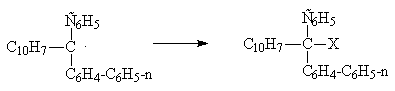
Несмотря на малые углы вращения, нет никакого сомнения в том, что оптическая активность возникла именно в результате действия циркулярно-поляризованного света; доказательством служит тот факт, что при перемене на обратный знак поляризации используемого света меняется на обратный и знак вращения продукта.
Е. Ферментативный асимметрический синтез.
Выделяемые из клеток ферменты — биологические катализаторы — способны осуществлять асимметрические реакции с высокой стереоспецифичностью, часто во много раз превосходящей стереоселективность обычных «химических» асимметрических синтезов. Например, фермент, выделяемый из мускульной ткани или из печени, способен катализировать асимметрическую внутримолекулярную реакцию Канниццаро:
CH3-CO-CHO  CH3-CHOH-COOH
CH3-CHOH-COOH
Сделаны и первые успешные попытки использовать ферменты для проведения асимметрического синтеза в лабораторных условиях; для этого используются так называемые иммобилизованные (закрепленные на полимерном носителе) ферменты.
Подчеркнем в заключение еще раз, что, как и в случае расщепления рацематов, оптически активные соединения генерируются только под действием оптически активных реагентов (катализаторов, растворителя, поляризованного света и т.п.).
Сервер создается при поддержке Российского фонда фундаментальных исследований
Не разрешается копирование материалов и размещение на других Web-сайтах
Вебдизайн: Copyright (C) И. Миняйлова и В. Миняйлов
Copyright (C) Химический факультет МГУ
Написать письмо редактору
Источник