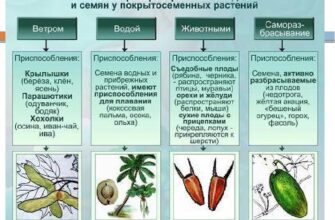
Методы выделения и разделения компонентов нефти

Методы выделения и разделения компонентов нефти.
Обнаружение и количественное определение воды, механических примесей в нефти. Метод Дина-Старка. Высокозастывающие компоненты нефти. Выделение и определение содержания в нефти нейтральных смол, асфальтенов, асфальтогеновых кислот по Маркуссону. Способы выделения и определения твердого парафина. Вымораживание (низкотемпературная кристаллизация) парафина. Методы клатрато — и комплексообразования. Фракционирование компонентов нефти с помощью клатратов мочевины и тиомочевины.
Мы приступаем к рассмотрению большого раздела физико-химических методов исследования нефти – методам выделения и разделения веществ.
Нефть сложная многокомпонентная система. Многие ее составные части близки по свойствам друг к другу. Для того чтобы выявить эти вещества и установить их концентрации необходимо выделить их из общей смеси или, хотя бы, сконцентрировать, чтобы уменьшить мешающее влияние других веществ.
Именно для этих целей и разработан комплекс физических и химических методов выделения и разделения компонентов нефти. Химические методы основаны на разной реакционной способности разделяемых компонентов. Физические – на разном распределении компонентов в двух или более равновесных фазах
Физические методы разделения компонентов нефти
Диффузия через мембрану
Диффузия с газом-носителем
Перегонка и ректификация
Перегонка с водяным паром
Диффузия через мембрану
Часть из них – простые методы, позволяющие полно отделить одни вещества от других, и уже не представляет труда определить их количество, применяя простое взвешивание или определение объема. Есть сложные методы с применением дополнительных разделяющих агентов (адсорбентов, комплексообразователей, селективных растворителей и т. д.), увеличивающих различие фаз.
Большинство этих методов мы рассмотрим в данном курсе.
С помощью физических методов из нефти выделяют воду, мех примеси, асфальтены, парафины и некоторые другие.
Стандартный метод определения воды в нефти – метод Дина и Старка (ГОСТ 2477-65).
Сущность метода. Вода отгоняется из нефти со специальным растворителем и ее объем измеряется в специальном приемнике. В качестве растворителя берут специальный обезвоженный прямогонный бензин 80-1200С или бензол.
Навеску нефти – 100 г или такую, чтобы выделившейся воды было не более 10 мл – помещают в круглодонную колбу и приливают 100 мл растворителя. Для равномерного кипения в колбу бросают касочки фаянса или стекла. К колбе присоединяют на шлифе насадку-ловушку (Дина-Старка) и обратный холодильник, как показано на рис. Верхний конец холодильника прикрывают ватой, чтобы внутрь не попала атмосферная влага.

Колбу нагревают до кипения и ведут перегонку так, чтобы отгон возвращался в колбу со скоростью 2-4 капли в сек. Вода стекает в приемник-ловушку. Перегонку прекращают как только уровень воды в ловушке перестанет увеличиваться. Обычно это время 30-60 мин. Минимальный определяемый объем воды этим методом – 0,03 мл.
Записывают объем воды с точностью до 1 деления и рассчитывают ее содержание в % мас. на нефть:
V – объем воды в приемнике-ловушке, мл
а – навеска нефти, г
Количественное определение мех. примесей по ГОСТ 6370-59.
Механические примеси в нефти выделяют фильтрованием и определяют весовым методом:
Навеску нефти растворяют в бензоле (и спиртоэфире) и фильтруют через бумажный фильтр. После высушивания фильтр взвешивают и по привесу определяют количество механических примесей.
Бумажные фильтры предварительно промывают тем растворителем, в котором будут растворять нефть и высушивают до постоянной массы при 105-1100С.
Берут навеску нефти от 100 до 25 г (в зависимости от вязкости), помещают в стакан и растворяют в бензоле при 1000С в 2-4 раза (при вязкости до 20 сСт) и в 4-6 раз (при вязкости выше 20 сСт).
Горячий раствор фильтруют через фильтр, укрепленный в воронке на штативе. Остаток в стакане смывают горячим бензолом на фильтр. Если в нефти есть вода, то остаток смывают на фильтр спиртоэфиром.
Фильтр с осадком промывают горячим бензолом пока фильтрат не будет прозрачным, а на фильтре не останется следов нефти.
Иногда полезно проводить такое фильтрование под вакуумом, используя воронку Бюхнера или воронку для горячего фильтрования (высоковязкие и парафинистые образцы).
Фильтр с осадком сушат при 105-1100С 30 мин и взвешивают. Операцию высушивания и взвешивания повторяют, пока вес не стабилизируется.
Рассчеты: M = 100[(m1-m2)/m3] , где
m1, m2 и m3 – масса фильтра с примесями, масса фильтра и навеска нефти, в г.
Содержание механических примесей до 0,005 % включительно рассматривается как их отсутствие.
Высокозастывающие компоненты нефти.
К высокозастывающим компонентам нефти относят парафины, смолы, асфальтены и асфальтогеновые кислоты.
Смолы, асфальтены и асфальтогеновые кислоты – это высокомолекулярные гетероатомные соединения нефти, объединяемые общим термином «смолисто-асфальтеновые вещества».
Смолы – вязкие или твердые, но легкоплавкие вещества (ММ=1000 и более). Содержание в нефтях от долей % до 25% в высокосмолистых. Асфальтены – твердые кристаллические вещества (ММ=10000 и более), при нагревании не плавятся, но переходят в пластичное состояние, а при более 3000 разлагаются. Количество в нефти менее 1%, в тяжелых нефтях и битумах – до 5%. Асфальтогеновые кислоты – смолоподобные вещества неопределенной струтуры. Содержатся в нефтях в следовых количествах. Обычно выделяются с асфальтенами и отделение от асфальтенов не проводят.
Общими для них методами выделения и концентрирования являются осаждение, соосаждение, кристаллизация и адсорбционный хроматографический метод.
Осаждение – метод разделения веществ, в процессе которого происходит образование новой фазы – осадка малорастворимого соединения. Эффективность разделения характеризуется отношением
К = m1/m0, где m0 – исходное количество осаждаемых веществ в растворе
m1 – часть перешедшая в осадок
Если в осадок переходит 99,9% вещества, осаждение считают полным.
В основе осаждения асфальтенов лежит их нерастворимость в легких парафинах (С5-7). Вернее другое их свойство: их молекулы – это ассоциаты пачек плоских параллельно расположенных молекул. Они растворимы в нефти и не слипаются, пока их окружает сольватная оболочка из других полярных молекул. Легкие алканы растворяют сольватную оболочку и они выпадают в виде грубодисперсного порошка.
Смолы и асфальтогеновые кислоты соосаждаются вместе с асфальтенами.
Причины соосаждения различны, обычно выделяют:
- Адсорбционное соосаждение – поглощение полярных молекул смол полярной поверхностью осадка асфальтенов. Адсорбция уменьшается при повышении температуры и при уменьшении адсорбирующей поверхности Окклюзия – захват смол всей массой осадка, не только его поверхностью. Причиной окклюзии является образование химических связей между осадком асфальтенов и молекулами смол и механический захват. Адсорбированные молекулы смол как бы зарастают новыми слоями асфальтенов. Послеосаждение – явление постепенного высаждение смол на уже сформировавшемся осадке в процессе отстаивания раствора.
Парафины, которые как смолисто-асфальтеновые вещества относятся к высокозастывающим компонентам нефти. Начиная с гексадекана С16 парафины уже при комнатной температуре находятся в твердом состоянии. При высоком содержании парафинов нефть теряет подвижность имеет мазеобразную консистенцию.
Выделить их можно низкотемпературной кристаллизацией. При понижении температуры из нефтяного раствора в первую очередь начинают выкристаллизовываться (вымораживаться) парафины.
Процедура осаждения и кристаллизации смол, асфальтенов и парафинов достаточно проста. В России существует стандартная методика на их определение в нефти – ГОСТ 11851-85 Нефть. Метод определения парафина.
Сущность метода заключается в предварительном удалении из нефти смолисто-асфальтеновых веществ, экстракции и удалении смол и последующем выделении парафинов смесью ацетона и толуола при минус 200С.
1. Навеска нефти 3-5 г разбавляют 40 кратным объемом н-гептана и выдерживают 16 ч. для полного осаждения асфальтенов. Асфальтены и соосадивщиеся с ними смолы и парафины отфильтровывают.
2. Фильтр с осадком сворачивают в пакет и экстрагируют смолы и парафины горячим н-гептаном в специальном экстракторе (см. рис)
3. экстракт смол и парафинов и фильтрат объединяют, отгоняют растворитель и начинают адсорбционное удаление смол. Для этого использют колонку с силикагелем (см. рис) нефть без асфальтенов вносят в колонку и промывают бензином и толуолом пока из колонки не будет стекать чистый растворитель.
4. Из фильтрата опять отгоняют растворитель и взвешиванием определяют массу обессмоленной и деасфальтенизированной нефти.

5. Заливают смесью ацетона и толуола 35:65 осаждают парафины в колбе в специальной охлаждающей бане. Быстро переносят кашицу парафинов на охлажденный фильтр и фильтруют под вакуумом

6. Затем парафины расплавляют и смывают растворителем в отдельную колбу, отгоняют растворитель и взвешивают.
Видно, что даже такая простая методика сложна, трудоемка, требует полного внимания химика и опыта проведения многостадийных работ.
Другими распространенными методами выделения парафинов являются методы комплексообразования.
В 1940 г. немецкий исследователь установил, что алифатические УВ с линейной структурой (н-алканы) начиная с гексана образуют кристаллические комплексы с карбамидом (мочевиной). Разветвленные алканы, циклоалканы и ароматические УВ почти не способны образовывать комплекс с мочевиной.
Карбамид – полярное, сильно ассоциированное соединение за счет действия межмолекулярных водородных связей.

Сам по себе карбамид кристаллизуется в тетрагональную кристаллическую решетку. При образовании комплекса с УВ решетка изменяется на гексагональную. Причем молекулы расположены по спирали на гранях правильных шестигранных призм.

Внутри спирали образуется канал гексагональной формы диаметром 0,525 нм. Поперечное сечение молекул н-алканов 0,42 нм, поэтому они хорошо вписываются в канал и удерживаются в нем за счет сил Ван-Дер-Ваальса. Молекулы разветвленных алканов, циклоалканов и ароматических УВ имеют диаметры превышающие эффективный диаметр алканов, поэтому они не способны образовывать аддукты с карбамидом.
Известно три типа аддуктов:
— Твердые комплексы, образующиеся в результате сильных донорно-акцепторных взаимодействий
— аддукты туннельного типа с полостями в кристаллической решетке, где могут помещаться молекулы УВ
— клатратные соединения с полостями в кристаллической решетке в виде клеток
В случае с карбамидом мы имеем дело с туннельными полостями. Стабильность комплекса возрастает с удлинением цепи алкана, а при длине неразветвленноой цепи выше С9 могут удерживаться и часть изоалканов.
С повышением температуры комплексы становятся менее устойчивыми, поэтому низкая температура более благоприятна для процесса их образования.
Количество комплексообразователя, которое требуется для выделения парафинов подбирается экспериментально. Считается, что на каждую СН2 группу требуется 0,7 моль карбамида.
Подобным комплексообразующим действием обладает тиокарбамид NH2C(S)NH2 – тоже туннельный комплексообразователь. Образует каналы большего диаметра (0,6-0,7 нм). Н-алканы в таких тунелях не удерживаются, зато «гостем» оказываются изоалканы, циклоалканы и некоторые арены. Тиокарбамид менее селективный комплексообразователь. Он позволяет лишь сконцентрировать отдельные группы веществ в аналитических целях.
Навеску нефтепродукта помещают в коническую колбу и разбавляют растворителем (бензол, хлороформ и др.) в соотношении 1:1, если высоковязкие вещества 1:5 или 1:10. Твердые фракции немного подогревают Добавляют порошок карбамида в соотношении 1:2 (керосиновые) или 1:4 (масляые) перемешивают и добавляют активатор – этиловый спирт 2,5 кратное количество по отношению к карбамиду. Активатор добавляют порциями при энергичном перемешивании. Если сразу добавить много – образуются корочки комплекса, затрудняюшие обработку. перемешивают во встряхивателе 1 час. И фильтруют осадок через фильтр Шота. Промывают осадок растворителем. Высушивают Комплекс разрушают несколькими порциями горячей дистиллированний воды при 60-800С Выделившиеся парафины извлекают из воронки гексаном с помощью делительной воронки (объяснить)
Определение солей в нефти обычно сводится к определению хлористых солей (хлорид-иона) методом титрометрии. Титрометрия наиболее редко применяемый метод при исследовании состава нефти. Поэтому теорию титрометрии мы рассматривать не будем, а только рассмотрим основные термины и законы, лежащие в ее основе.
- Титр – число граммов вещества в 1 мл раствора Титрованный раствор – раствор, содержащий точно измеренное количество реагента Титрование – постепенное прибавление титрованного раствора к анализируемому раствору для определения эквивалентного количества вещества Точка эквивалентности – момент, когда количество добавленного титранта химически эквивалентно количестве титруемого вещества. Эту точку отмечают по изменению индикатора
Источник
29 Разделение компонентов нефти
2. Методы разделения компонентов нефти
3. Использование методов разделения
в промышленном масштабе
Рекомендуемые файлы
Вступление. Что же такое нефть? Теплотехник ответит, что это прекрасное, высококалорийное топливо. Но химик возразит: нет! Нефть – это сложная смесь жидких углеводородов, в которых растворены газообразные и другие вещества. И чтобы перечислить все продукты, получаемые из нефти, нужно потратить несколько листов, так как их уже несколько тысяч.
Еще Д.И. Менделеев заметил, что топить печь нефтью все равно, что топить ее ассигнациями.
Историческая справка. Дмитрий Иванович Менделеев (1834-1907). Русский химик, открывший периодический закон химических элементов, разносторонний ученый, педагог и общественный деятель. Получил образование на отделении естественных наук физико-математического факультета Главного Педагогического Института в Петербурге, курс которого окончил в 1855 г. с золотой медалью. Защитил множество магистерских и докторских диссертаций, читал лекции в качестве доцента. Среди его трудов – фундаментальный работы по химии, химическим технологиям, физике, метрологии, воздухоплаванию, сельскому хозяйству, экономики, народному просвещению. Написал труд «Основы химии». В 1869 г открыл периодический закон химических элементов.
Нефть (от перс. neft) — горючая маслянистая жидкость со специфическим запахом, распространенная в осадочной оболочке Земли и являющаяся важнейшим полезным ископаемым.
В настоящее время нефтехимия дает почти четверть всей химической продукции. Нефть – ценнейшее природное ископаемое, открывшее перед человеком удивительные возможности «химического перевоплощения». Всего производных нефти насчитывается уже около 3 тысяч.
Нефть занимает ведущее место в мировом топливно-энергетическом хозяйстве. Ее доля в общем потреблении энергоресурсов непрерывно растет. Нефть составляет основу топливно-энергетических балансов всех экономически развитых стран.

МЕТОДЫ РАЗДЕЛЕНИЯ КОМПОНЕНТОВ НЕФТИ
С необходимостью разделения смеси веществ на составляющие ее компоненты приходится сталкиваться как химику-синтетику, химику-аналитику, так и технологу, геологу, физику, биологу и многим другим специалистам. Особое значение разделение смеси веществ приобрело в последние десятилетия в связи с проблемой получения сверхчистых веществ.
Разделение смеси не вызывает особых трудностей, если ее компоненты находятся в различных фазах. Оно существенно осложняется, если компоненты смеси образуют одну фазу. В этом случае приходится изменять агрегатное состояние отдельных компонентов (например, добиться их выпадения в осадок), либо применять химические или физические методы разделения. В основе последних лежат кинетические явления или фазовые равновесия.
Такие широко известные методы разделения, как дистилляция, кристаллизация, экстракция и адсорбция основаны на изменении фазовых равновесии. В этих процессах молекулы веществ, образующих смесь, переходят через границу раздела, стремясь к такому распределению между фазами, при котором в каждой из них устанавливается постоянная равновесная концентрация.
Если свойства компонентов исследуемой смеси близки, то достаточная степень разделения достигается лишь многократным повторением элементарного акта разделения. Такой процесс, например, осуществляется в насадочных или тарельчатых ректификационных колоннах. Следует отметить, что в таких случаях полное разделение возможно только для простых (не более чем трехкомпонентных) систем.
Более полного разделения можно достичь, если на эффект, вызываемый многократным установлением фазовых равновесий, наложить действие кинетического фактора. В тех случаях, когда используются кинетические явления (например, при молекулярной дистилляции), через поверхность раздела фаз и лишь в одном направлении переносятся молекулы только одного вещества. Если разделение смеси производить в таких системах, в которых одна из фаз (подвижная) перемещается относительно другой (неподвижной), то улавливание и удаление молекул, покидающих поверхность раздела фаз, осуществляется благодаря постоянному перемещению подвижной фазы. Как и при фазовом равновесии, молекулы, выходящие из подвижной фазы, возвращаются в нее, попадая, однако, не в прежний элемент ее объема, а в новый.
Если в процессе разделения фазовые переходы повторять многократно, то можно получить высокую эффективность разделения. Так как фазовые переходы связаны с поверхностью раздела, подвижная и неподвижная фазы должны обладать большой поверхностью соприкосновения. Кроме того, вследствие наличия диффузионных процессов, снижающих эффективность разделения, обе фазы должны иметь относительно небольшую толщину взаимодействующего слоя.
В виду сложности химического состава нефти для разделения ее на более или менее однородные группы и фракции применяются самые разнообразные методы: перегонка (дистилляция) и ректификация, адсорбция – десорбция, экстракция, кристаллизация, получение твердых комплексных соединений и некоторые другие.
Братья Дубинины впервые создали устройство для перегонки нефти. С 1823 г. Дубинины стали вывозить фотоген (керосин) многими тысячами пудов из Моздока внутрь России. Завод Дубининых был очень прост: котел в печке, из котла идет труба через бочку с водой в пустую бочку. Бочка с водой – холодильник, пустая – приемник для керосина.
В Америке впервые опыты по перегонке нефти осуществил в 1833 г. Силлиман.
Нефть — как уже было указано, представляет собой чрезвычайно сложную смесь, взаимнорастворимых углеводородов. Разделить ее нацело на составляющие компоненты практически невозможно, но этого для промышленного применения нефтепродуктов и не требуется. В промышленной практике нефть делиться на фракции, различающиеся температурными коэффициентами. Это разделение проводиться на установках первичной перегонки нефти с применением дистилляции и ректификации.
Получение фракции служит дальнейшим сырьем для переработки или используется как товарные продукты. Первичная перегонка – первый технологических процесс перегонки нефти. Установки первичной перегонки имеются на каждом нефтеперерабатывающем заводе.
Перегонка под давлением и под вакуумом. Простая перегонка не может дать удовлетворительного разделения жидкостей с близкими температурами кипения. Поэтому она применяется только для грубого разделения на широкие фракции. Так, например, при химическом групповом анализе бензины и керосины разделяются на стандартные фракции 60-95 0 С, 95-122 0 С, 122-150 0 С и т.д. перегонкой с дефлегматором. Разгонка при температурах выше 300 0 С проводится под вакуумом во избежание термического разложения высокомолекулярных углеводородов. С этой целью применяют разгонку с водяным паром или в струе инертного газа, чаще всего азота.
Перегонка с ректификацией — самый распространенный способ точного фракционирования по температурам кипения. Принцип работы лабораторных ректификационных установок заключается в том, что пары жидкости из колбы или куба отводятся не в конденсатор, как при простой перегонке, а поступают в ректификационную колонку, чаще всего насадочного типа. Поднимаясь по колонке, пары достигают верха и оттуда поступают в дефлегматор-конденсатор, где они конденсируются. Полученный конденсат частично отбирается через холодильник в приемник, но большая его часть вновь попадает в колонку, стекая по насадке сверху вниз. Эта часть конденсата называется флегмой. Таким образом, в колонке образуются два потока: нагретые пары движутся снизу вверх, а охлажденная жидкая флегма – сверху вниз. Между жидкой и паровой фазами по всей высоте колонки происходит интенсивный теплообмен. В результате нагретые пары испаряют из жидкой фазы наиболее летучие компоненты, а более холодная флегма конденсирует из паров наименее летучие составные части. При этом теплота конденсации нижележащего слоя используется для испарения жидкости вышележащего слоя. Следовательно, жидкость и пар в результате многократного повторения процессов испарения и конденсации все время обмениваются компонентами. Этот процесс можно уподобить последовательному повторению процесса простой перегонки с загрузкой в колбу каждый раз отгона от предыдущей перегонки. Очевидно, что при разгонке бинарной смеси в результате ректификации пар наверху колонки, а следовательно, и отбираемый конденсат, будет сильно обогащен низкокипящим компонентом, а пар внизу колонки, а следовательно, и жидкость в колбе, наоборот, — высококипящим. При ректификации многокомпонентной смеси, например бензина, отбор можно производить через любые интервалы температуры и получать, таким образом, узкие фракции с небольшим количеством компонентов. Четкость погоноразделения лабораторных ректификационных колонок зависит от многих факторов. Большое значение имеют материал и форма насадки, которая должно обладать сильно развитой поверхностью, на ней и происходит соприкосновение паров с флегмой. Чем лучше качество насадки, тем меньше высота, эквивалентная одной теоретической тарелке (ВЭТТ). С этой величиной, понятно, связана и высота колонки. Не менее важное значение имеет правильно выбранное флегмовое число, т.е. отношение объема флегмы к объему отбора за одинаковый промежуток времени, а также скорость отбора дистиллята. Четкость ректификации зависит, кроме того, от диаметра колонки и других конструктивных особенностей, а также от соблюдения адиабатичности по всей высоте колонки, т.е. иначе говоря, от тщательности тепловой изоляции.
Эффективность лабораторных колонок принято оценивать числом теоретических тарелок в рабочих условиях (ЧТТ). В зависимости от состава перегоняемых смесей на практике используются колонки с ЧТТ от 20 до 150 и выше. Так, например, подсчитано, что для получения 40% дистиллята от загрузки с содержанием в нем 95 % низкокипящего компонента для смеси гептан — толуол с разницей температур кипения, равной 12,4 0 С, необходима колонка, эквивалентная только 10 теоретическим тарелкам, а для смеси гептан – изооктан с температурой кипения, равной 0,8 0 С, уже – 150 теоретическим тарелкам.
Лабораторные ректификационные установки применяются для самых различных целей. На них можно разгонять при низких температурах с помощью жидкого азота или твердой углекислоты смеси газообразных углеводородов. При атмосферном давлении ректифицируются смеси, выкипающие в интервале 30-200 0 С. И, наконец, под вакуумом разгоняют на узкие фракции высококипящие погоны нефти
3. ИСПОЛЬЗОВАНИЕ МЕТОДОВ РАЗДЕЛЕНИЯ В ПРОМЫШЛЕННОМ МАСШТАБЕ
Использование дистилляционных методов разделения компонентов нефти в промышленном масштабе происходит: на нефтепромыслах отделяют попутные нефтяные газы, механические примеси, воду (до остаточного содержания 0,5-1% по массе) и растворенные в ней минеральные соли (100-1800 мг/л), на нефтеперерабатывающих заводах глубоко обезвоживают (не более 0,2%) и обессоливают (не более 3 мг/л), а легкое сырье одновременно стабилизируют – выделяют пропан — бутановую, а иногда и пентановую фракции углеводородов для снижения их потерь при перегонке нефти.

Принципиальная технологическая схема установки для атмосферно-вакуумной перегонки нефти. Аппараты 1, 3 – атмосферные ректификационные колонны; 2 — печи для нагрева нефти и мазута; 4 — вакуумная ректификационная колонна; 5 – конденсаторы – холодильники; 6 – теплообменники.
Линии: I – нефть; II – легкий бензин; III – отбензиненая нефть; IV – тяжелый бензин; V – керосин и газойль; VI – водяной пар; VII – мазут; VIII – газы разложения; IX – масляные фракции; Х – гудрон.
Перегонка может быть осуществлена однократным, многократным или постепенным испарением. При однократном испарении в течение всего времени нагревание смеси продуктов до определенной конечной температуры образующиеся пары не выделяются из системы и остаются в контакте с жидкостью. После того как сообщение тепла заканчивается, вся парожидкостная смесь вводится в сепаратор. Здесь образовавшиеся пары в один прием отделяются от жидкости.
При многократном осуществлении процесса разделения фаз производится несколько приемов. Многократное испарение состоит из повторяющейся несколько раз процесса однократного испарения. Первоначально происходит отделение паров от жидкости, а затем – на второй ступени – жидкая фаза, оставшаяся при отделении паров в первой ступени, вновь испаряется и т.д.
При постепенном испарении образующиеся пары по мере их образования непрерывно вводятся из перегонного аппарата. Постепенное испарение применяется при лабораторной перегонке нефти из колбы, а промышленной практике прежде использовалось при перегонке на кубовых установке.
Процесс однократного испарения обладает преимуществами перед постепенным испарением. При однократном испарении низкокипящие фракции, пройдя в пары, остаются в аппарате, снижают парциальное давление испаряющихся высококипящих фракций, что дает возможность вести перегонку при более низких температурах.
При постепенном испарении, наоборот, легкие фракции отгоняют сначала, а тяжелые – в конце. Поэтому легкие фракции, которые превратились в пары и были выведены из аппарата, не влияют на температуру кипения тяжелых фракций. Благодаря влиянию легких фракций, применяя однократное испарение, можно снизить конец кипения перегоняемого сырья на 50-100 0 С по сравнению с постепенным испарением.
На современных установках перегонки нефти проводятся с применением однократного испарения. Как известно в составе нефти имеются углеводороды, кипящие при атмосферном давлении в интервале температур 400-500 0 С и выше, в то время как термическая стабильность углеводородов сохраняется только до 380-400 0 С. при более высокой температуре начинается процесс разложения – крекинга углеводородов, причем наиболее высококипящие углеводороды нефти обладают наименьшей термической стабильностью.
Для того чтобы избежать разложения углеводородов надо понизить температуры их кипения. Это достигается перегонкой нефти под вакуумом. Нефтяная фракция, выкипающая при атмосферном давлении в интервале температур 450-500 0 С, может быть перегнана под вакуумом (остаточное давление 20-40 мм рт.ст.) при 200-250 0 С.
Для понижения температуры кипения в практике нефтепереработки применяют также перегонку с водяным паром, который снижает парциально е давление углеводородов.
Понизить температуры кипения фракции можно и перегонкой с инертным газом (азот, углекислый газ и т.д.).
Однако этот метод не нашел распространения, так как присутствие инертного газа ухудшает условия конденсации нефтяных фракций.
На современных установках первичной перегонки нефти применяю совместные действия пониженного давления и ввода водяного пара.
При однократном испарении взаимно растворимых жидкостей и последующей конденсации паров получают две фракции: легкую, в которой содержится больше низкокипящих фракций, и тяжелую, в которой содержится меньше низкокипящих фракций, чем в исходном сырье. Следовательно, при перегонке происходит обогащение одной фазы низкокипящими, а другой – высококипящими компонентами. Однако достичь требуемого разделения компонентов нефти и получить конечные продукты, кипящие в заданных температурных интервалах, с помощью перегонки нельзя. Поэтому после однократного испарения нефтяные фракции подвергаются ректификации.
Ректификацией называется диффузионный процесс разделения жидкостей, различающихся по температурам кипения, за счет противоточного, многократного паров и жидкости.
Контактирование паров и жидкости осуществляется в вертикальных цилиндрических аппаратах – ректификационных колоннах, снабженных специальными устройствами – ректификационными тарелками или насадками, — позволяющими создать тесный контакт между паром, поднимающимся вверх по колонне и жидкостью, стекающей вниз.
Средняя часть в виде пара, жидкости или парожидкостной смеси подается сырье, которое необходимо разделить на две части – высококипящей и низкокипящей. В простейшем случае исходное сырье состоит из двух компонентов (например, бензола, толуола, бутана, изобутана и др.). однако чаще сырье представляет собой многокомпонентную смесь, которую с помощью ректификации надо разделить на два продукта, один из которых содержит в основном низкокипящие компоненты, а другой – высококипящие.
Зона, в которую подается сырье, носит название эвапорационной, так как в ней происходит эвапорация – однократное испарение нагретой в печи или теплообменнике смеси на паровую и жидкую фазы. В некоторых случаях эвапорационная зона отделена от колонны, эвапорация производится в самостоятельном аппарате. Однако у большинства колонн, в частности на установках первичной перегонки, однократное испарение и ректификация совмещаются.
Принцип работы промышленной ректификационной колонны аналогичен лабораторной. В работающей ректификационной колонне через каждую тарелку проходит четыре потока: 1) жидкость – флегма, стекающая с выше лежащей тарелки, 2) пары, поступающие с ниже лежащей тарелки, 3) жидкость – флегма, уходящая на ниже лежащую тарелку, 4) пары, поднимающиеся на выше лежащую тарелку.
Пары и жидкости, поступающие на тарелку не находятся в состояния равновесия, однако, вступая в соприкосновение, стремятся к этому состоянию. Жидкий поток с вышележащей тарелки поступает в зону более высокой температуры, и поэтому из него испаряется некоторое количество низкокипящего компонента, в результате чего концентрация последнего в жидкости уменьшается. С другой стороны, паровой поток поступающей с нижележащей тарелки, попадает в зону более низкой температуры и часть высококипящего продукта из этого потока конденсируется, переходя в жидкость. Конденсация высококипящего компонента пара, таким образом, понижается, а низкокипящего – повышается. Фракционный состав жидкости по высоте колонны непрерывно изменяется.
Часть ректификационной колонны, которая расположена выше ввода сырья, называется концентрационной, а расположенная ниже ввода – отгонной. В обеих частях колонны происходит один и тот же процесс ректификации. С верха концентрационной части в паровой фазе вводится целевой продукт необходимой чистоты – ректификат, а с низа – жидкость, все еще достаточной степени обогащенная низкокипящим компонентом. В отгонной части из этой жидкости окончательно отпаривается низкокипящий компонент. В виде жидкости с низа этой части колонны вводится второй целевой компонент – остаток.
Для нормальной работы ректификационной колонны необходимо, чтобы с верха колонны на ниже лежащей тарелки непрерывно стекала жидкость (флегма). Поэтому часть готового продукта (ректификата) после конденсации возвращается на верхнюю тарелку колонны в виде так называемого орошения. С другой стороны для нормальной работы колонны необходимо, чтобы с низа колонны вверх непрерывно поднимались пары. Чтобы создать в колонне паровой поток, часть уходящего из колонны остатка подогревается, испаряется и возвращается обратно в колонну.
Ректификационные колонны. В зависимости от внутреннего устройства колонны делятся на тарелочные и насадочные. На большинстве технологических установок современного нефтеперерабатывающего завода применяются только тарелочные колонны. Существуют ректификационные тарелки различных типов – колпачковые, бесколпачковые, струйно-направленные и др.
Колпачковая тарелка представляет собой металлический диск, в котором имеется множество отверстий для прохода паров. По периметру отверстий закреплены бортики определенной высоты, называемые стаканами, благодаря которым на тарелке поддерживается определенный слой жидкости. Сверху стаканы накрываются колпачками. Между верхним срезом стакана и колпачком имеется зазор для прохода паров, поступающих с нижележащей тарелки. При работе колпачки погружены в слой жидкости, и вследствие этого образуется гидравлический затвор, через который барботирует пары.
Уровень жидкости на тарелках поддерживается сливными перегородками (сливными карманами), нижняя часть которых доходит до следующей тарелки. Избыток жидкости по сливным карманам спускается на нижележащую тарелку. Положение колпачком можно регулировать, изменяя размеры зазора между колпачком и верхним срезом стакана. Очень важно, чтобы тарелки размещались по колонне строго горизонтально и чтобы все колпачки были одинаково погружены в жидкость на тарелке. Если эти требования не выполнены, то в какой-либо части тарелки толщина слоя жидкости будет меньше. Через эту часть тарелки начнет проходить большое количество жидкости, и многие колпачки на остальной части тарелки перестанет работать.
Наиболее распространены колпачковые тарелки желобчатого типа, тарелки с S-образными элементами, с круглыми колпачками и тарелки клапанного типа.
Желобчатые тарелки имеют простую конструкция и весьма легко монтируются. Тарелка представляет собой прямоугольник или квадрат, вписанный в поперечное сечение колонны. Один из сегментов, отделяемых этим прямоугольником служит сливным устройством данной тарелки, другой – сливным устройством вышележащей. Два сегмента тарелки – глухие.
Тарелка состоит из нескольких желобов, прикрепленных к опорным уголкам. Над желобами располагаются колпачки, монтируемые на нужной высоте. Жидкость движется по тарелке вдоль колпачков. Основной недостаток желобчатых тарелок заключается в малой площади барботажа (до 30% от площади тарелки), что способствует увеличению скорости паров и уноса флегмы.
В отличие от желобчатых тарелок в тарелках с S-образными элементами жидкость, направляясь к сливному устройству, движется поперек колпачков, а сами колпачки представляют одно целое с желобом. Каждый S-образный элемент состоит из колпачковой и желобчатой части. При сборке их располагают таким образом, чтобы колпачковая часть одного элемента перекрывала желобчатую часть другого, образуя гидравлический затвор.
Тарелки из S-образных элементов предназначены для колонн, работающих при атмосферном или невысоком давлении, для них характерно устойчивое равномерная работа при изменении нагрузок. Производительность тарелок на 20% больше, чем желобчатых.
Еще более эффективны для колонн, работающих при переменных нагрузках по пару и жидкости, а также для колонн, в которых требуется добиться повышенной четкостью разделение, клапанные прямоточные тарелки. Основной элемент такой тарелки – клапан, который под действием паров приподнимается над полотном тарелки на различную высоту. В отличие от прочих колпачковых тарелок, работающих в статичном режиме, для клапанных тарелок характерен динамический переменный режим работы.
Подвижные клапаны в зависимости от паровой нагрузки поднимаются или опускаются, регулируя площадь свободного сечения тарелки. Благодаря такой конструкции, в широком пределе нагрузок, определенном возможной длиной хода клапана, скорость паров существенно не меняется.
Из бесколпачковых тарелок применение в последние годы нашли решеточные тарелки провального типа и ситчатые тарелки с отбойными элементами.
Основным показателем для тарелок с переливами являются скорость паров в свободном сечении колонны. Скорость паров в колоннах установок первичной перегонки зависит от типа тарелки, расстояние между тарелками, нагрузки тарелки по жидкости, физических свойств разделяемых продуктов и других факторов. Она составляет: в атмосферной колонне 0,6-0,9 м/с, в отбензинивающей — 0,2-0,3 м/с, в стабилизаторе 0,15-0,2 м/с, в вакуумной – 2-3 м/с.
В данной работе приведен расчет тарельчатой ректификационной колонны для разделения бинарной смеси бензол — толуол.
4. ОСОБЕННОСТИ РАСЧЕТА ТАРЕЛЬЧАТОЙ РЕКТИФИКАЦИОННОЙ КОЛОННЫ
Как правило, расчет ректификационной колонны производится для заданных: составе исходной смеси, кубового остатка, дистиллята, производительности и рабочем давлении в колонне.
В начале определяется материальный баланс колонны и рабочее флегмовое число. Для этого используется диаграмма y -x . Затем подбирается тип тарелок, определяется скорость пара, диаметр колонны, коэффициенты массопередачи, высота колонны, гидравлическое сопротивление тарелок. После этого можно провести расчет эксплуатационных свойств, а также экономические показатели ее использования.
4.1. Пример расчета ректификационной колонны для перегонки смеси бензол — толуол
Для примера, рассчитаем колонну при содержании легколетучего компонента (т.е. бензола) в исходной смеси 35%(масс.), в дистилляте 98%, в кубовой жидкости 1,7%. Производительность по исходной смеси 5кг/с.
4.2. Материальные расчеты
4.2.1 Материальный баланс колонны
Производительность по дистилляту P и кубовому остатку W определяется из уравнения материального баланса ректификационной колонны:

 (1)
(1)

 (2)
(2)
 (3)
(3)
Все расчеты в данном случае ведутся для легкокипящего компонента, а значит х есть концентрация бензола. Для дальнейших расчетов необходимо пересчитать составы фаз из массовых в мольные по соотношению :
 (4)
(4)
где x — мольная доля компонента А, 
 — массовая доля компонента А, % (масс.)
— массовая доля компонента А, % (масс.)
МА — мольная масса компонента А, 
МВ — мольная масса компонента В,
Подставив мольные массы бензола и толуола получаем:



4.2.2. Определение рабочего флегмового числа
Нагрузки ректификационной колонны по пару и жидкости определяются значением рабочего флегмового числа R. Флегмовое число являет собой отношение количества флегмы к количеству дистиллята. Оно может находиться в интервале от Rmin до ¥. При минимальном флегмовом числе можно получить максимальное количество дистиллята, но число тарелок становится бесконечно большим. Если флегмовое число принять равным бесконечности, то получится, что колонна работает сама на себя. При флегмовом числе меньше минимального мы ни при каких условиях не сможем получить конечный продукт с заданными свойствами.

Вообще флегмовое число отражает угол наклона рабочей линии к оси абсцисс для верхней части колонны и входя в уравнение рабочей линии. Уравнение рабочей линии для верхней части колонны выглядит как:
 (5)
(5)
yD , как впрочем и yW определяются равными xD и xW соответственно. Иначе говоря предполагается что состав паровой и жидкой фазы одинаков как для низа так и для верха колонны. Все это можно увидеть на диаграмме 1.
Минимальное флегмовое число определяется по следующей формуле:
 (6)
(6)
где  — мольная доля спирта в паре, находящемся в равновесии с исходной смесью, определяется по y-x диаграмме.
— мольная доля спирта в паре, находящемся в равновесии с исходной смесью, определяется по y-x диаграмме.

Рабочее значение флегмового числа примем равным 2,1. Для определения рабочего флегмового числа существует множество рекомендаций, мы их упускаем, но их можно найти в [3].
4.2.3. Построение рабочей линии на диаграмме “жидкость — пар”.
Рабочая линия процесса ректификации, в отличие от процесса абсорбции, представляет собой совокупность рабочих линий для верхней и для нижней части колонны и характеризуется изломом в точке соответствующей составу питательной смеси.
Для верхней части колонны можно воспользоваться уравнением (5), а для нижней части колонны существует уравнение:
 (6)
(6)
Вид рабочей линии представлен на все том же рисунке 1.
4.2.4 Определение среднего массового расхода по жидкости
Средние массовые расходы по жидкости для верхней и нижней частей колонны определяются из соотношений:
 (7)
(7)
 (8)
(8)
где МP и МF — мольные массы дистиллята и исходной смеси,
МВ и МН — мольные массы жидкости в верхней и нижней частях, .
Мольная масса жидкости в верхней и нижней частях колонны соответственно
равна :
 (9)
(9)
 (10)
(10)
где Мб и Мт — мольные массы бензола и толуола
xср.в и xср.н — средний мольный состав жидкости соответственно в верхней и нижней частях колонны:




Аналогично находится мольная масса исходной смеси:

Мольную массу дистиллята можно принять равной мольной массе бензола.
Подставив результаты соотношения в (7) (8) получаем:


4.2.5 Определение среднего массового расхода по пару
Средние массовые потоки пара в верхней и нижней частях колонны
соответственно равны :
 (11)
(11)
 (12)
(12)
где  и
и  — средние мольные массы паров в верхней и нижней частях колонны:
— средние мольные массы паров в верхней и нижней частях колонны:
 (13)
(13)
 (14)
(14)
где средние значения состава паровой фазы рассчитываются аналогично жидкой фазе и равны:


Тогда из формул (13) и (14) следует


Подставив результаты в (11) (12) получаем:


4.3 Скорость пара и диаметр колонны
На этой стадии необходимо выбрать тип тарелки. Поскольку предполагается, что жидкость не содержит взвешенных частиц выберем используем ситчатые тарелки.
Допустимая скорость в верхней и нижней части колонны определяется по формуле:
 (15)
(15)
Поскольку плотности бензола и толуола близки, то плотность жидкой фазы может быть приближенно определена как 796 кг/м 3 .
Средняя плотность пара для нижней и для верхней части колонны может быть определена по формуле:
 (16)
(16)
где t — температура для верхней или для нижней части колонны.
Температура в колонне, в свою очередь, определяется по диаграмме t — x,y, которую можно увидеть на диаграмме 2.

По средним составам фаз определим температуру в верхней части колонны 89°С, в нижней части колонны 102°С.
Тогда по формуле (16) рассчитываем плотность паровой фазы соответственно в нижней и верхней части колонны.



Сейчас можно рассчитать допустимые скорости как в верхней, так и в нижней части колонны:



Диаметр колонны может быть определен по формуле:
 (17)
(17)
Диаметр колонны принимается одинаковым по всей ее высоте и как правило равен большему из определенных. Однако, в данном случае различия между скоростями в верхней и нижней части колонны не велики поэтому можно использовать средние значения:



Подставив их в формулу (17) получим:
 м
м
Приняв стандартный размер обечайки равным 1,8м уточним рабочую скорость пара. Она будет равной 0,82м/с.
На данном этапе необходимо выбрать тарелку из ряда стандартных. Опуская процесс выбора, отметим, что это тарелка ТС-Р с ниже приведенными характеристиками:
Источник